甲醇在金红石型TiO2(110)表面的吸附解离
2014-11-14孙秀明樊红军周丹红
孙秀明,王 煦,樊红军,周丹红
(1. 辽宁师范大学 化学化工学院, 辽宁 大连 116029; 2. 中国科学院 大连化学物理研究所,辽宁 大连 116023)
金红石型二氧化钛TiO2具有稳定性好和催化活性高的特点,因此越来越多地受到人们的关注。无论是在理论上还是实验上,金红石型二氧化钛在光催化中都有许多应用,如太阳能电池、水的分解、传感器和生物材料等[1,2],但是近年来金红石型二氧化钛更多的研究是将其作为催化剂或光催化剂[3-11],尤其是有机分子光降解和水的分解[5,12,13],这些在环境整治和洁净能源方面有很重要的意义。1980年,Sato和White研究得出结论纯净的二氧化钛对水分解制H2没有显著地催化活性[14],同年Kawai和Sakata研究发现在水中加入甲醇能显著地增加 H2的产生[15,16]。因此,需要在分子水平上理解甲醇在 TiO2模型表面上的光化学作用,这有助于提高开发新的、有效的分解水的光催化剂的能力。但是甲醇在二氧化钛表面的存在方式尚不明确,这为催化理论研究提出了挑战。1997年Bates、Kresse和Gillan运用基于第一性原理的密度泛函理论(DFT)-赝势方法系统地计算了TiO2(110) 表面能和结构[17],他们选取了2到7层的TiO2(110)表面结构,并选择了不同的真空宽度进行计算。他们认为,只要真空宽度在4Å以上,真空宽度的变化则对表面结构影响不大,对于简单的要求来说四层足够了,但是六层或七层是得到表面能收敛的最好层数。1998年Bates和Gillan又对甲醇以分子和解离两种状态吸附在 TiO2(110)表面进行了研究[18],他们先用五层的金红石型 TiO2(110) 表面做静电计算,再用三层的模型进行分子动力学模拟。研究结果发现:甲醇的解离状态能更为稳定地吸附;在静电计算中得出通过C-O键断裂和通过O-H键断裂从能量基础上是等同的,但是他们在分子动力学模拟中并没有发现C-O键断裂,因此他们认为甲醇分子在 TiO2(110)表面上是以 O-H键断裂形式解离的。 2007年R. Sa nchez de Armas等人运用第一原理计算了甲醇在TiO2(110) 表面上的吸附与解离[19]。他们用周期性都DFT方法、平面波基组和赝势计算了甲醇在完整的和有缺陷的TiO2(110) 表面上的吸附和解离情况,他们选择了4到6层的结构,他们的收敛值显示甲醇的解离态比分子态更稳定。2012年Guo等人用6层的TiO2研究了水和甲醇在TiO2表面的光催化解离[16],他们选择的是50%的吸附度,且解离方式是朝向不同的方向解离,在正文中他们并没有详细讨论结构的变化。人们对于金红石型TiO2(110) 表面的研究越来越多,但是在理论计算中所选取的模型却是不同的,因此得到不同的结果。本文将选择较大的6层模型和较小的吸附度(25%)来考察甲醇在不同的 TiO2(110) 表面模型上的吸附与解离。
1 实验部分
本文计算使用基于平面波密度泛函理论的VASP(Vienna Ab initio Simulation Package)[20,21]软件包。交换相关能用广义梯度近似(GGA)方法中的PBE(Perdew, Burke and Ernzerhof)[22]泛函来计算。电子和离子相互作用通过缀加投影波 PAW(projector augmented wave)函数来描述。Kohn-Sham单电子采用平面波基组展开,截断能设为400 eV,布里渊区采用4×1×2的Monkhorst-Pack格点[23]描述。
本文使用4×1的周期平板模型来研究CH3OH在TiO2(110)表面的吸附和解离行为。我们选择金红石型TiO2(110) 表面(即在金红石型晶体的a、b、c三维空间里分别选 a=1,b=1,c=0,即 a轴、b轴所在平面)并截取模型,如图 1所示。在(110) 表面上有五配位钛和六配位钛,有二配位的桥氧和三配位的面氧。
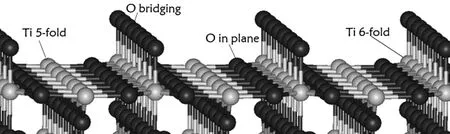
图1 金红石型二氧化钛(110)表面(红色(深色)为氧原子,灰色(浅色)为钛原子)Fig.1 The rutile TiO2 (110) surface, the red globes are represented as O atoms, the grey ones are represented as Ti atoms
根据前人的计算结果[16-20],我们选择六层的二氧化钛4×1模型,该模型是由6层Ti原子和6层O原子组成。为了消除层与层之间的相互作用,真空层高度设为10 Å。对所截取的二氧化钛模型分别进行饱和固定(Model A)和饱和放开(Model B)处理。饱和固定是将下表面的五配位钛用水分子饱和,使其成为体相中的六配位钛;将最底部的两层原子固定,其余原子全部放开优化。饱和放开是将下表面的五配位钛用水分子饱和,使其成为体相中的六配位钛;所有原子全部放开优化。在这两个模型上,分别研究甲醇分子以氧原子吸附[10]在五配位钛上的吸附、解离和过渡态的三种状态,通过对甲醇分子的吸附能、解离能和过渡态能垒的分析比较可以得到甲醇在金红石型二氧化钛表面的存在形式。同时,考察不同模型对计算结果的影响。
甲醇分子的吸附能计算公式如下:

式中E1是甲醇分子在TiO2模型上的稳定吸附物的总能量,E(TiO2)是优化的TiO2模型的总能量,E(MeOH) 是甲醇分子优化后的总能量。
甲醇分子的解离能计算公式如下:

式中E3是甲醇分子在TiO2模型上的稳定解离物的总能量,E1是甲醇分子在TiO2模型上的稳定吸附物的总能量。
甲醇分子的过渡态能垒计算公式如下:

式中E2是甲醇分子在TiO2模型上的过渡态的总能量,E1是甲醇分子在TiO2模型上的稳定吸附物的总能量。
2 结果与讨论
图2和图3分别给出是饱和固定模型和饱和放开模型上优化后的甲醇分子吸附、解离和过渡态的结构和几何结构参数。
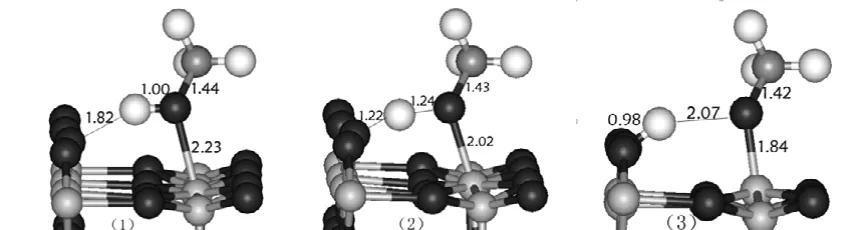
图2 饱和固定6×4×1模型TiO2 (110) 表面上甲醇分子吸附解离过程中各键长(单位为Å)变化(红色球、浅灰色球、白色球、深灰色球分别代表O、Ti、H、C原子)Fig.2 The changes of bond lengths of methanol molecule on the TiO2(110) surface of the saturated froze 6×4×1 model during the dissociation process, the red, gray and white spheres represent O atoms, Ti atoms and H atoms,respectively.
在饱和固定模型上,甲醇分子首先以分子形式吸附在二氧化钛表面的五配位钛上,此时甲醇的羟基氧与五配位钛之间的距离为2.23 Å,表明形成配位键,羟基氢与桥氧之间的距离为1.82 Å,形成氢键。在过渡态,羟基的O-H键增长了0.24 Å,羟基氢原子与甲氧基逐渐脱离,向其对面的桥氧运动,距离缩小了0.60 Å,同时甲醇的羟基氧与五配位钛之间距离减小了0.21 Å。最后羟基氢原子到达桥氧上,与桥氧形成稳定的O-H键,其键长为0.98 Å,与此同时,甲氧基与五配位钛的距离为1.84 Å,形成稳定的离子键。在三种状态中甲氧基的C-O键长变化很小,从1.42到1.44 Å。

图3 饱和放开6×4×1模型TiO2 (110) 表面上甲醇分子吸附解离过程中各键长(单位为Å)变化(红色球、浅灰色球、白色球、深灰色球分别代表O、Ti、H、C原子)Fig.3 The changes of bond lengths of methanol molecule on the TiO2(110) surface of the saturated relaxed 6×4×1 model during the dissociation process, the red, gray and white spheres represent O atoms, Ti atoms and H atoms,respectively.
在饱和放开模型上,甲醇分子吸附的状态与饱和固定模型上的相同。但是在过渡态,羟基的O-H键更长而羟基氢原子与对面的桥氧更靠近,解离后,断裂的氢与甲醇的氧距离更远。
能量计算的结果列于表1,图4是反应过程的能线图。从反应能线图可以看出,甲醇首先吸附在二氧化钛表面上,都是放热过程,这可能是由于甲醇的氧上孤对p电子与表面五配位钛的空的d轨道配位作用,同时,甲醇与表面桥氧生成了氢键也有利于吸附。在两个模型上的解离反应能垒分别是4.54和4.45 kcal/mol, 吸附能和解离能也都只有很小差别,这是在DFT计算误差范围内的,说明两种模型对计算结果没有很大影响。从理论计算结果来看,甲醇吸附和解离过程都是放热过程,而且解离所克服的能垒也很低。分子形式吸附状态下的能量E1和解离形式吸附状态下的能量E2非常接近,初步认为,在二氧化钛表面上,两种形式都是存在的,解离态略微稳定。以上计算结果与文献18和19提到的结论一致。人们在实验中会看到分子吸附形式更稳定[10],可能是由于吸附度比较大的原因。

表1 甲醇、二氧化钛模型及甲醇在模型表面上各种存在形式的优化结果Table 1 Energies of optimization of methanol, TiO2 and kinds of methanol on TiO2 model surface

图4 甲醇在不同模型上TiO2 (110) 表面吸附解离能线图Fig.4 The adsorption dissociation energy profile of methanol on different TiO2 surface model
3 结 论
本文运用理论计算方法研究了甲醇在金红石型二氧化钛(110)表面的吸附方式。用周期性的从头算方法研究甲醇分子吸附在金红石型二氧化钛(110)表面的吸附解离情况,从而确定出甲醇在金红石型二氧化钛(110)表面的吸附方式。首先在二氧化钛110表面上截取六层的4×1模型,进行饱和固定和饱和放开处理,再模拟出一个甲醇以氧原子吸附在模型表面的五配位钛上的吸附、解离和过渡态模型,然后通过对甲醇分子在金红石型二氧化钛(110)表面的吸附解离前后的结构优化得到吸附能、解离能和过渡态的能垒的分析和各键长的分析比较,得到甲醇在金红石型二氧化钛模型表面的吸附方式。研究结果表明,甲醇在金红石型二氧化钛表面有分子吸附和解离吸附两种吸附方式,但以解离吸附方式为主。
[1]Antonio T,Selloni A. Methanol Adsorption and Reactivity on Clean and Hydroxylated Anatase(101) Surfaces[J].. J. Phys. Chem. B. ,2004,108(50):19314-19319.
[2]Diebold U. The surface science of titanium dioxide[J]. Surf. Sci.Rep.,2003,48(5-8):53-229.
[3]Wahlström E,Vestergaard E K,Schaub R. Electron Transfer-Induced Dynamics of Oxygen Molecules on the TiO2(110) Surface[J].Science,2004,303(511):511-513.
[4]Gratzel M. Photoelectrochemical cells. Natrue.2001,414(6861):338-344.
[5] Fox M A,Dulay M T. Heterogeneous Photocatalysis[J]. Chem. Rev.,1993,93(1):341-357.
[6]Linsebigler A L,Lu G,Yates J T, Jr. Photocatalysis on TiOnSurfaces:Principles, Mechanisms, and Selected Results[J]. Chem. Rev. ,1995,95(3):735-758.
[7]Khan S U, Al-Shahry M, Ingler W B,Jr. Efficient Photochemical Water Splitting by a Chemically Modified n-TiO2[J]. Science, 2002,297(2240):2243-2245.
[8]Wang R,Hashimoto K, Fujishima A et al. Light-induced amphiphilic surfaces[J].Natrue,1997,388(6641):431-432.
[9]Kamat P V. Photochemistry on Nonreactive and Reactive (Semiconductor)Surfaces[J]. Chem.Rev.,1993,93(1):267-300.
[10]Zhang Z,Bondarchuk O,White J M et al. Imaging Adsorbate O-H Bond Cleavage: Methanol on TiO2(110)[J]. Chem.Soc.,2006,128(13):4198-4199.
[11]Chen X,Liu L,Yu P Y et al. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals[J]. Science,2011,331(746):746-750.
[12]Hoffmann M R,Martin S T,Choi W et al. Environmental Applications of Semiconductor Photocatalysis[J]. Chem. Rev. ,1995,95(1):69-96.
[13]Ollis D F,Alekabi H. Photoreactors for Purification and Decontamination of Air. 1ST International Conf on TiO(2) Photocatalytic Purification and Treatment of Water and Air[M]. London, Canada .1992:481-494.
[14]Sato S,White J M. Photodecomposition of water over Pt/TiO2catalysts[J].Chem. Phys. Lett., 1980,72(1):83-86.
[15]Kawai T,Sakata T J. Photocatalytic Hydrogen Production from Liquid Methanol and Water[J]. Chem.Soc.,Chem.Commum., 1980,24(15):694-695.
[16]Guo Q,Xu C,Ren Z et al. Stepwise Photocatalytic Dissociation of Methanol and Water on TiO2(110)[J]. J. Am. Chem. Soc., 2012,134(32):13366-13373.
[17]Bates S P,Kresse G,Gillan M J. A systematic study of the surface energetics and structure of TiO2(110) by first-principles calculations[J]. Surface Science, 1997,385(2-3):386-394.
[18]Ramamoorthy M,Vanderbilt D. First-principles calculations of the energetics of stoichiometric TiO2surfaces[J]. Phys. Rev. B., 1994,49(23):16721-16728.
[19]Bates S P,Gillan M J. Adsorption of Methanol on TiO2(110): A First-Principles Investigation[J]. J. Phys. Chem. B., 1998,102(11):2017-2026.
[20]Kresse G,Hafner J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B.1993,48(17):558-561.
[21]Kresse G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996,54(16):11169-11186.
[22]Perdew J P,Burke K,Emzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. B. 1996,77(18):3865-3868.
[23]Monkhorst H J,Pack J D. Special points for Brillonin-zone integrations.Phys. Rev. B. 1976,13(12):5188-5192.
