CTAB-NaOH混合液碱处理HZSM-35分子筛催化DME羰基化反应
2014-10-22刘小行刘盛林李秀杰谢素娟徐龙伢赵明军
刘小行,刘盛林,李秀杰,谢素娟,徐龙伢,曾 蓬,赵明军
(1.中国科学院 大连化学物理研究所,辽宁 大连116023;2.中国科学院大学,北京100049;3.中国寰球工程公司 辽宁分公司,辽宁抚顺113006;4.中国石油 抚顺石化分公司,辽宁 抚顺113004)
乙酸甲酯是一种具有应用前景的脂肪酸酯[1],其传统生产工艺存在腐蚀设备、污染环境、生产成本高的缺陷,因此,其绿色、廉价的合成方法备受关注。Cheung等[2]首次报道,具有八元环孔道结构的分子筛催化剂可实现二甲醚(DME)无卤、非贵金属催化羰基化制备乙酸甲酯,且其八元环内的B酸位是羰基化反应的活性位[3-4]。Liu等[5-6]的研究表明,在催化DME羰基化制备乙酸甲酯反应中,酸性丝光沸石在低温下表现出很高的催化活性和产物选择性,但其失活速率较快;ZSM-35分子筛催化剂则表现出较好的稳定性,但其催化活性相对较低。采用NaOH碱处理,可使HZSM-35催化剂产生一部分介孔,明显提高其催化活性和稳定性[7-8]。Schmidt等[9]采用十六烷基三甲基溴化铵(CTAB)和NaOH混合溶液对ZSM-5分子筛进行碱处理,使其在甲醇制烯烃反应中具有良好的稳定性;Yoo等[10]采用CTAB和NaOH混合溶液碱处理的方法,得到了具有双介孔结构并具有高结晶度的ZSM-5分子筛。
笔者利用不同浓度CTAB和NaOH混合溶液对HZSM-35分子筛进行碱处理,并借助N2吸附-脱附、Py-IR、TPO表征手段,研究该催化剂的DME羰基化反应催化性能与其物理化学性质的关系,以期对该催化反应有更深入的了解。
1 实验部分
1.1 原料
硝酸铵、氢氧化钠(AT),分析纯,天津市科密欧化学试剂开发中心产品;十六烷基三甲基溴化铵(CTAB),分析纯,百灵威公司产品;二甲醚(DME),分析纯,大连光明特种气体有限公司产品;一氧化碳(CO),分析纯,大连光明特种气体有限公司产品。ZSM-35分子筛原粉,n(Si)/n(Al)=14.0,上海卓悦化工科技有限公司产品。
1.2 催化剂制备
将ZSM-35分子筛原粉进行铵交换,得到HZSM-35分子筛粉末(HZ35)。将CTAB与NaOH配成混合碱溶液,使其中NaOH的浓度在0.1~1.0mol/L范围,并且 CTAB与NaOH摩尔比在0.1~2.0范围。以6倍于HZ35质量的混合碱溶液将HZ35浸泡其中,80℃水热处理4h。离心分离,水洗至中性,120℃干燥10h,550℃焙烧6h。用1.0mol/L硝酸铵溶液在85℃下交换3次,经水洗、干燥、540℃焙烧3h、成型,即得HZSM-35催化剂,记为xAT-yCTAB,其中x代表NaOH浓度,y代表CTAB浓度。
1.3 催化剂表征
采用帕纳克公司X Pert Pro型X射线衍射仪进行催化剂的晶相分析,CuKα射线,管电压40kV,管电流40mA。假设HZ35催化剂的结晶度为100%,由xAT-yCTAB与HZ35二者的XRD谱在2θ为 9.4°±0.1°、22.4°±0.1°、22.7°±0.1°、23.3°±0.1°、23.7°±0.1°、24.5°±0.1°和25.3°±0.1°处衍射峰峰高之和的比值得到xAT-yCTAB催化剂的相对结晶度(RC)。采用飞利浦公司 MagiX型X射线荧光光谱仪进行元素分析,铜靶,通过IQ+New无标定量软件计算获得数据。采用麦克公司ASAP2020型物理吸附仪表征催化剂的孔结构。采用程序升温脱附装置获得样品的NH3-TPD曲线[11]。采用布鲁克公司VERTEX70型傅里叶变换红外光谱仪进行催化剂的Py-IR表征[12]。采用自制程序升温氧化装置和巴尔查斯公司Omistar型质谱仪测定催化剂积炭。称取80mg催化剂置于U形石英管中,在流量为35mL/min O2/Ar(V(O2)/V(Ar)=1/10)混合气流中,以10℃/min的速率从室温升至850℃,在线检测m/e=28(CO)信号变化(TPO曲线)。
1.4 催化剂评价
采用常规的固定床反应器评价催化剂催化性能。反应管内径16mm、长33cm,催化剂(20~40目)装量7g。催化剂在N2气氛下520℃预处理2h,然后冷却至反应温度。原料为DME和CO混合物,自上而下通过催化剂床层进行反应。采用安捷伦公司7890A型气相色谱仪在线分析反应产物组成,PONA色谱柱,FID检测器。
2 结果与讨论
2.1 CTAB-NaOH混合碱液浓度对HZSM-35分子筛物理化学性质的影响
图1为不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的XRD谱。由图1可见,所有被测样品都具有FER结构的特征衍射峰。当混合碱液中CTAB浓度为0.40mol/L时,随着其中NaOH浓度从0增加到0.80mol/L,碱处理HZSM-35的结晶度先升高后降低,但都高于未经碱处理的HZ35。这是因为适当条件下碱处理可以脱除HZSM-35中的无定形硅铝物种,相当于对分子筛晶体进行了纯化[13]。1.00mol/LNaOH的混合碱液处理得到的1.00AT-0.40CTAB的相对结晶度下降至66%,表明分子筛微孔结构遭到了部分破坏。当混合碱液中NaOH浓度为0.40mol/L、CTAB 浓度不同时,碱处理HZSM-35的相对结晶度比HZ35催化剂均有一定程度的提高;而0.40AT-0.04CTAB、0.40AT-0.40CTAB和0.40AT-0.80CTAB的相对结晶度均低于0.40AT,这是因为碱洗下来的一部分硅铝物种通过CTAB的作用,返沉积在分子筛晶体表面所致[9-10,14]。
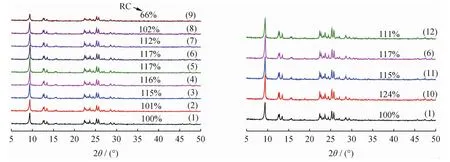
图1 不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的XRD谱Fig.1 XRD patterns of HZSM-35zeolites alkaline-treated by CTAB-NaOH solutions with different concentrations
图2为不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的N2吸附-脱附等温线。由此计算所得各样品的孔结构数据与相应n(Si)/n(Al)值一并列于表1。
由图2可见,HZ35的N2吸附-脱附等温线偏离了典型的I型等温线,在相对压力p/p0=0.8处开始出现滞回环,这是由于HZ35分子筛是片状晶体团簇体,具有较多二次堆积介孔[7]。当混合碱液中CTAB浓度为0.40mol/L时,随着其中NaOH浓度从0增至0.60mol/L,碱处理 HZSM-35的N2吸附-脱附等温线并无明显的变化,特别是未出现晶内介孔;而NaOH浓度为0.80和1.00mol/L混合碱液所得碱处理HZSM-35的N2吸附-脱附等温线在p/p0=0.45处开始出现滞回环。由表1可见,0.80AT-0.40CTAB 和 1.00AT-0.40CTAB 的外比表面积明显高于HZ35,且二者的N2吸附-脱附等温线在p/p0=0.45处开始出现滞回环,表明高NaOH浓度混合碱液所得碱处理HZSM-35出现了一定量的晶内介孔[7]。此外,0.40AT、0.40AT-0.04CTAB 和 0.40AT-0.80CTAB3个碱处理HZSM-35的N2吸附-脱附等温线与HZ35的相比也无明显变化。

图2 CTAB-NaOH混合溶液碱处理HZSM-35分子筛的N2吸附-脱附等温线Fig.2 N2adsorption-desorption isotherms of HZSM-35zeolites alkaline-treated with CTAB-NaOH solutions
由表1还可知,与 HZ35相比,0.10~0.60mol/L NaOH混合碱溶液处理后分子筛的微孔比表面积均有不同程度的增加,这是因为低NaOH浓度碱溶液处理的主要作用是部分脱除孔道内的无定形硅物种,与XRD结果一致;但1.00mol/L NaOH混合碱溶液处理后的1.00AT-0.40CTAB微孔比表面积降至223m2/g,低于HZ35,表明其微孔结构遭到破坏。当混合碱溶液的NaOH浓度为0.40mol/L时,随着其中CTAB浓度的升高,碱处理HZSM-35分子筛的微孔比表面积逐渐降低,但都高于HZ35。另外,碱处理HZSM-35的微孔体积的变化规律与其微孔比表面积的相似。
表1同时显示,当混合碱溶液的CTAB浓度为0.40mol/L时,随着其中NaOH浓度从0增加到1.00mol/L,碱处理 HZSM-35的n(Si)/n(Al)下降。低 NaOH 浓度 (0~0.40mol/L)碱处理HZSM-35的n(Si)/n(Al)比 HZ35的高,可能是因为硅物种通过CTAB的作用返沉积在分子筛晶体上的量大于铝物种所致[9-10,14]。当混合碱溶液的NaOH浓度为0.40mol/L时,随着其中CTAB浓度的升高,碱处理 HZSM-35的n(Si)/n(Al)逐渐升高,再一次证明了CTAB的返沉积作用。

表1 不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的n(Si)/n(Al)及孔结构数据Table 1 n(Si)/n(Al)and pore properties of HZSM-35zeolites alkaline-treated by CTAB-NaOH solutions with different concentrations
图3为不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的NH3-TPD曲线。由图3可见,所有被测样品的NH3-TPD曲线形状相似,在250℃和520℃各出现1个脱附峰,分别对应弱酸和强酸中心[7],表明CTAB和NaOH混合溶液碱处理不改变HZSM-35的酸类型,但影响其总酸量。当混合碱溶液的CTAB浓度为0.40mol/L时,随着其中NaOH浓度从0增加至0.20mol/L,碱处理HZSM-35分子筛的总酸量低于HZ35;NaOH 浓度在0.40~0.80mol/L 范围,碱处理HZSM-35分子筛的总酸量明显高于HZ35;进一步提高NaOH浓度到1.00mol/L,碱处理 HZSM-35分子筛骨架部分坍塌,结晶度下降,总酸量降低。当混合碱溶液的NaOH浓度为0.40mol/L时,随着其中CTAB浓度的增加(0~0.80mol/L),碱处理HZSM-35的总酸量逐渐增加,并且都高于HZ35。
2.2 CTAB-NaOH混合碱液浓度对碱处理HZSM-35分子筛催化DME羰基化反应性能的影响
在n(CO)/n(DME)=10、T=235℃、p=2.0MPa、MHSV(DME)=0.13h-1条件下,不同浓度 CTABNaOH混合溶液碱处理HZSM-35分子筛催化DME羰基化反应的DME转化率随时间的变化示于图4。HZ35催化DME羰基化反应过程分为诱导期、稳定期和衰减期3个阶段,诱导期中DME转化率逐渐升高,然后进入稳定期,最后进入衰减期[7]。由图4(a)可见,采用相同浓度CTAB、不同浓度NaOH混合溶液碱处理的HZSM-35在催化DME羰基化反应诱导期内的DME转化率变化规律性不强,衰减期内的变化也不明显。0.40CTAB催化DME反应的转化率和MA选择性与HZ35相比几乎没有差别,说明单独CTAB碱处理并未改变催化剂的活性;随着混合碱溶液中NaOH浓度从0.10mol/L增至0.80mol/L,碱处理 HZSM-35催化DME反应的稳定期中DME转化率先升高后降低。与HZ35催化剂相比,0.10AT-0.40CTAB的催化活性明显提高,诱导期阶段DME初始转化率由34.2%升至53.7%,稳定期阶段DME转化率由50.0%升至56.0%;随着反应的进行,HZ35的催化活性下降,31h时 DME转化率降至45.9%,而0.10AT-0.40CTAB的DME转化率仍然保持在52.2%,呈现出良好的活性稳定性。在碱处理HZSM-35催化剂中,0.20AT-0.40CTAB催化DME反应稳定期的DME转化率为最高,达到64.4%,31h时为 61.6%;0.80AT-0.40CTAB催化剂尚无诱导期,随着反应的进行,DME转化率迅速下降,31h时已降至47.6%;1.00AT-0.40CTAB的稳定期DME转化率降为41.8%。
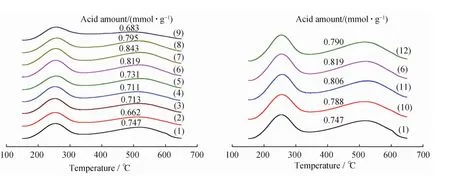
图3 CTAB-NaOH混合溶液碱处理HZSM-35分子筛的NH3-TPD曲线Fig.3 NH3-TPD profiles of HZSM-35zeolites alkaline-treated with CTAB-NaOH solutions

图4 不同浓度CTAB-NaOH混合溶液碱处理的HZSM-35催化DME羰基化反应的DME转化率随时间的变化Fig.4 DME conversion vs time in DME carbonylation over HZSM-35alkali-treated by CTAB-NaOH solutions with different concentrations
由图4(b)可见,与HZ35相比,0.40AT催化DME羰基化反应稳定期的DME转化率升至54.9%,31h时为52.0%,表明采用0.40mol/L NaOH溶液处理的 HZMSM-35的催化活性得到提高,与Li等[7]的结果一致;0.40AT-0.04CTAB催化DME羰基化反应稳定期的DME转化率达到62.4%,31h时保持在58.7%,表明在NaOH碱处理过程中添加CTAB,有助于进一步提高催化剂的活性;随着CTAB浓度从0.08mol/L增至0.80mol/L,碱处理催化剂催化DME羰基化反应稳定期的DME转化率无明显变化。
不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛催化DME羰基化反应的MA和甲醇(MeOH)选择性随时间的变化示于图5。
由图5(a)、(c)可见,反应0~12h期间,xAT-0.40CTAB(x分别为 0.10、0.40、0.60)和0.40AT-yCTAB(y分别为0、0.04、0.08、0.80)催化DME反应的MA选择性迅速增加,12~31h期间MA选择性进入稳定期,差别不大。0.15AT-0.40CTAB和 0.20AT-0.40CTAB 催 化DME反应的MA选择性在反应4h后即进入了稳定期,0.80AT-0.40CTAB催化剂的MA初始选择性较高,随着反应的进行,MA选择性先降低后升高,15h后进入稳定期,达到98%左右,1.00AT-0.40CTAB催化剂的 MA选择性迅速由92.5%(2h)降低至67.5%左右(19h),为最差。由图5(b)、(d)可见,DME反应甲醇选择性的变化趋势则与MA选择性的变化相反。由上可知,适当条件碱处理,可有效提高HZSM-35催化剂活性,同时使之具有良好的稳定性。

图5 不同浓度CTAB-NaOH混合溶液碱处理HZSM-35催化DME羰基化反应的MA和MeOH选择性随时间的变化Fig.5 MA and MeOH selectivity vs time in DME carbonylation over HZSM-35alkali-treated by CTAB-NaOH solutions with different concentrations
催化DME羰基化反应31h后的各碱处理HZSM-35催化剂的TPO曲线示于图6。由图6可见,将被测样品的TPO曲线拟合后均得到375、475、575和675℃4个积炭氧化峰,且峰形类似,说明CTAB-NaOH混合溶液碱处理不改变HZSM-35催化剂的积炭类型。将积炭HZ35催化剂的积炭氧化峰面积设为100%,可计算出其他积炭的碱处理HZSM-35催化剂的积炭氧化峰面积的相对大小,即相对积炭量。计算得到,积炭碱处理催化剂的相对积炭量均不超过100%,且0.80AT-0.40CTAB、0.40CTAB和0.10AT-0.40CTAB 催化剂的积炭量明显多于0.15AT-0.40CTAB 和0.60AT-0.40CTAB 催化剂,而0.40AT-0.04CTAB 和0.40AT-0.40CTAB催化剂的积炭量高于0.40AT-0.80CTAB催化剂。结合被测样品在催化DME羰基化反应中甲醇选择性的大小顺序(见图5)可发现,催化剂积炭量与甲醇选择性存在一定关系。甲醇选择性越高,催化剂积炭量越多,暗示其积炭主要来源于反应副产物甲醇。因为甲醇是一种很活泼的化合物,很容易在催化剂上形成积炭[5]。0.20AT-0.40CTAB催化剂的甲醇选择性较低,但是其积炭量较高,可能与该催化剂DME转化率较高有关;HZ35催化剂积炭量高于0.80AT-0.40CTAB催化剂,可能由于后者具有部分介孔所致。这样的说法均需进一步探讨。
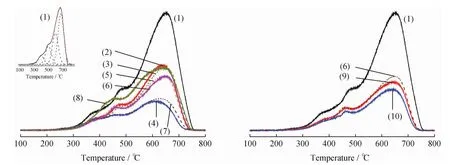
图6 催化DME羰基化反应31h后的碱处理HZSM-35催化剂的TPO曲线Fig.6 TPO profiles of the spent HZSM-35zeolite catalysts for 31hof DME carbonylation
TPO分析结果表明,CTAB-NaOH混合溶液碱处理HZSM-35分子筛,可有效抑制其上积炭的生成。Xue等[15]报道,通过减小丝光沸石的尺寸(由微米级减至纳米级),可有效地缩短反应物或产物达到或者脱离反应活性位的时间,从而提高DME转化率;更重要的是,抑制了硬积炭的生成,使催化剂具有较好的稳定性。
2.3 HZSM-35分子筛催化性能与其物理化学性质的关系
HZSM-35分子筛由十元环(0.42nm×0.54nm)和八元环(0.35nm×0.48nm)组成[16]。其中八元环内的B酸位被认为是DME羰基化的主要活性中心[2-4,17]。反应过程中,甲氧基在八元环内以平行于孔道轴线的方向被吸附,有利于CO攻击表面甲基生成过渡态CH3CO*物种,更重要的是,其较小的孔径可以稳定过渡态。吡啶分子动力学直径为0.54nm,与HZSM-35分子筛中十元环的孔径接近,但大于八元环孔径。在温度300℃、吡啶分压10Pa条件下,经过4600min接触,吡啶分子可以完全覆盖HZSM-35分子筛中孔道内的酸性位[18],而在相同温度下,经过短时间接触(30min),吡啶分子只能进入十元环孔道[12]。因此,在吸附温度300℃、吸附时间30min条件下,通过Py-IR可以得到吸附在HZSM-35分子筛外表面及十元环孔道内B酸(1540cm-1)和 L酸(1450cm-1)位的定量信息。这样,碱处理HZSM-35催化剂八元环中总酸量可用NH3-TPD测出的总酸量与Py-IR测出的分子筛外表面及十元环中总酸量的差值(ΔAcidsite)来量化。根据十元环中B酸与L酸酸量之比CB/L,笔者推测,催化剂中八元环中绝大部分酸为B酸。同时,引入分子筛催化剂介孔与微孔体积比VMeso/VMicro来描述孔道扩散性能。需要指出的是,ΔAcidsite应较为接近HZSM-35催化剂的八元环真实总酸量。笔者试图按van Donk等[19]所使用方法对HZSM-35催化剂八元环B酸量进行拟合量化,但是由于基线漂移,很难甚至不能拟合。微孔孔径与催化剂本身的结构相关,而介孔是微孔的堆积,因此,用VMeso/VMicro来描述分子筛催化剂孔道扩散性能具有一定的合理性。
CTAB-NaOH混合溶液碱处理HZSM-35分子筛的物理化学性质与其催化DME羰基化反应的转化率列于表2。由表2可见,HZSM-35的催化活性不仅与其八元环的总酸量有关,还与孔道扩散性能相关。分子筛的八元环总酸量太低,如HZ35,即使其催化DME羰基化反应的中间体及产物MA能及时从孔道中移出,但因八元环中活化的DME量少,因此DME转化率较低,即此时总酸量为反应的速率控制步骤。在合适的总酸量下,DME羰基化反应的中间体及产物在孔道中的扩散性能显得尤为突出[7,15]。因 此,xAT-0.40CTAB 催 化 剂 (x分别为0.15、0.20、0.40、0.60)的DME转化率高于xAT-0.40CTAB(x分 别 为 0.10、0.80、1.0)和0.40AT催化剂,其中,前者催化DME羰基化反应各DME转化率相近,是酸性和孔道扩散性能共同作用的结果。0.80AT-0.40CTAB和1.00AT-0.40CTAB虽然产生了晶内介孔,但微孔结构遭到破坏,总酸量降低,导致 DME转化率降低。0.40AT-0.04CTAB和0.40AT-0.40CTAB 的 ΔAcidsite值和VMeso/VMicro值均相近,因此其催化DME反应的转化率也相近。另外,在本研究条件下,催化剂的总酸量和孔道扩散性能对MA选择性的影响不甚明显。
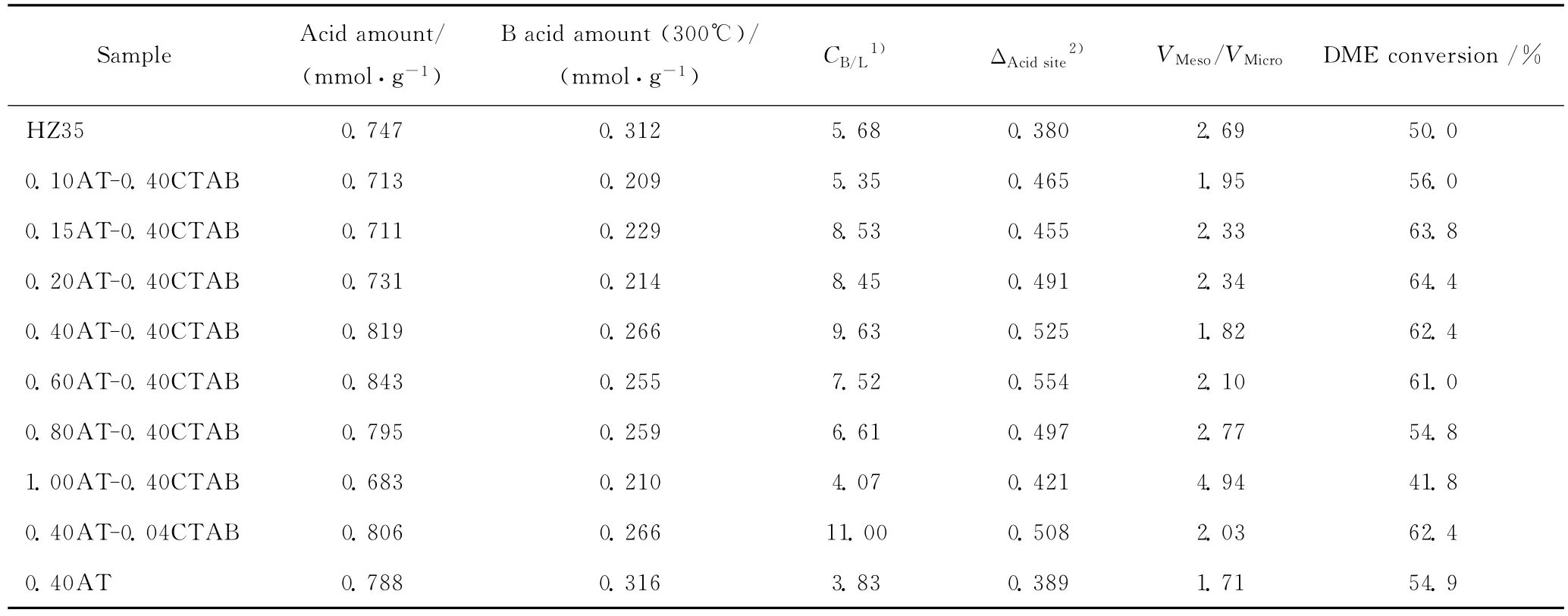
表2 不同浓度CTAB-NaOH混合溶液碱处理HZSM-35分子筛的物理化学性质及其催化DME羰基化反应的转化率Table 2 Physico-chemical properties of HZSM-35zeolites alkali-treated by CTAB-NaOH solutions with different concentrations and the DME conversion of DME carbonylation catalyzed by them
3 结 论
(1)0.40mol/L CTAB和0.60mol/L NaOH 混合溶液碱处理可较好地脱除HZSM-35分子筛孔道内的无定形硅铝物种,增加其总酸量,特别是八元环的总酸量。
(2)HZSM-35分子筛的DME羰基化催化活性与其八元环总酸量、孔道扩散性能有关。0.40mol/L CTAB和0.20mol/L NaOH混合溶液碱处理可使其具有较高的八元环总酸量和较好的孔道扩散性能,在n(CO)/n(DME)=10、T=235℃、p=2.0MPa、MHSV(DME)=0.13h-1条件下,其催化DME羰基化反应的转化率可高达64.4%。
(3)CTAB-NaOH混合溶液碱处理可有效抑制HZSM-35催化剂上积炭的生成。
[1]LEE J S,KIM J C,KIM Y G.Methyl formate as a new building block in C1chemistry[J].Applied Catalysis,1990,57(1):1-30.
[2]CHEUNG P,BHAN A,SUNLEY G J,et al.Selective carbonylation of dimethyl ether to methyl acetatecatalyzed by acidic zeolites[J].Angewandte Chemie-International Edition,2006,45(10):1617-1620.
[3]CHEUNG P,BHAN A,SUNLEY G J,et al.Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites[J].Journal of Catalysis,2007,245(1):110-123.
[4]BHAN A,ALLIAN A D,SUNLEY G J,et al.Specificity of sites within eight-membered ring zeolite channels for carbonylation of methyls to acetyls[J].Journal of the American Chemical Society,2007,129(16):4919-4924.
[5]LIU J L,XUE H F,HUANG X M,et al.Stability enhancement of H-mordenite in dimethyl ether carbonylation to methyl acetate by pre-adsorption of pyridine[J].Chinese Journal of Catalysis,2010,31(7):729-738.
[6]LIU J L,XUE H F,HUANG X M,et al.Dimethyl ether carbonylation to methyl acetate over HZSM-35[J].Catalysis Letters,2010,139(1):33-37.
[7]LI X J,LIU X H,LIU S L,et al. Activity enhancement of ZSM-35in dimethyl ether carbonylation reaction through alkaline modifications [J]. RSC Advances,2013,3(37):16549-16557.
[8]BONILLA A,BAUDOUIN D,PEREZ-RAMIREZ J.Desilication of ferrierite zeolite for porosity generation and improved effectiveness in polyethylene pyrolysis[J].Journal of Catalysis,2009,265(2):170-180.
[9]SCHMIDT F,LOHE M R,BUCHNER B,et al.Improved catalytic performance of hierarchical ZSM-5 synthesized by desilication with surfactants [J].Microporous and Mesoporous Materials,2013,165(1):148-157.
[10]YOO W C,ZHANG X Y,TSAPATSIS M,et al.Synthesis of mesoporous ZSM-5zeolites through desilication and re-assembly processes[J].Microporous and Mesoporous Materials,2012,149(1):147-157.
[11]许国梁,朱向学,刘盛林,等.ITQ-13分子筛的表面改性及其丁烯催化裂解性能[J].石油学报(石油加工),2009,25(增 刊 ):28-31. (XU Guoliang,ZHU Xiangxue,LIU Shenglin,et al.Catalytic performances of surface modified ITQ-13zeolies in butene cracking[J]. Acta Petrolei Sinica (Petroleum Processing Section),2009,25(Suppl):28-31.)
[12]KHITEV Y P,IVANOVA I I,KOLUAGIN Y G,et al.Skeletal isomerization of 1-butene over micro/mesoporous materials based on FER zeolite[J].Applied Catalysis A:General,2012,441–442(0):124-135.
[13]van LAAK A N C,GOSSELINK R W,SAGALA S L,et al.Alkaline treatment on commercially available aluminum rich mordenite[J].Applied Catalysis A:General,2010,382(1):65-72.
[14]VERBOEKEND D,VILE G,PEREZ-RAMIREZ J.Mesopore formation in USY and beta zeolites by base leaching:Selection criteria and optimization of poredirecting agents[J].Crystal Growth & Design,2012,12(6):3123-3132.
[15]XUE H F,HUANG X M,EVERT D.Coking on micrometer-and nanometer-sized mordenite during dimethyl ether carbonylation to methyl acetate[J].Chinese Journal of Catalysis,2013,34(8):1496-1503.
[16]BORADE R B,CLEARFIELD A.Synthesis of ZSM-35 using trimethylcetylammonium hydroxide as a template[J].Zeolites,1994,14(6):458-461.
[17]BHAN A,IGLESIA E.A link between reactivity and local structure in acid catalysis on zeolites[J].Accounts of Chemical Research,2008,41(4):559-567.
[18]PIETERSE J A Z,VEEFKINGD P S,SESHAN K,et al.On the accessibility of acid sites in ferrierite for pyridine[J].Journal of Catalysis,1999,187(2):518-520.
[19]van DONK S,BUS E,BROERSMA A,et al.Butene skeletal isomerization over H-ferrierite:A TEOM and in situ IR study on the role of carbonaceous deposits and the location of Br∅nsted acid sites[J].Applied Catalysis A:General,2002,237(1-2):149-159.
