苯分子与Si6O18H12和Al6O24H30团簇模型相互作用的理论研究
2014-10-18宋开慧
王 幸 钱 萍,* 宋开慧 张 超 宋 伟
(1山东农业大学化学与材料科学学院,山东泰安 271018;2泰安市疾病预防控制中心,山东泰安 271000)
1 Introduction
The monoaromatic hydrocarbons benzene,toluene,ethylbenzene,and xylenes are abbreviated by BTEX,which is commonly found in liquid and gas states in our planet,especially abundant in pollutants,present in soils,in industrial process as well as in the product of petroleum refining.BTEX is mainly produced in catalytic synthesis,the pyrolysis of naphtha,and oil refinement.1BTEX,playing an important role in the synthesis of other compounds,is the main pollutant to the soils and groundwater,and has resulted in great harm to the people because of its high toxicity,carcinogenicity,and mutagenicity.2-4BTEX migration is controlled by diffusion and adsorption on the surface of soil components,where the clay minerals are ubiquitous.For further understanding the surface structural and chemical properties of clay mineral,it is important to know the adsorption structures and interactions between the pollutants and clay surface at the molecular level.
To the best of our knowledge,the important crystal structure information of clay minerals has been determined by some available experimental techniques.5,6However,the particles of clay minerals are extremely small sizes and in structural and substitutional disorder for the natural material.These factors complicate an experimental study of structure and properties of clay.In particular,these small particles,which often prevent utilizing single-crystal diffraction for the precise determination of structural parameters,lead to ambiguous crystal structure.Therefore,special attention is given to the related study of clay systems at the molecular level,especially,the adsorbent properties of clay surfaces.7Essentially,two main approaches should be used in theoretical studies of clay.The first one is based on the exploitation of the translational symmetry in the calculation procedure,e.g.,periodic ab initio calculations,6,8which may be impractical or over costly for a very large system just like ours.The second one is based on the application of standard molecular approach in which the cluster is formed by cutting the periodic structure and the model is treated as a molecule.9-12However,it is well-known that the disadvantage of the cluster approach is the limited cluster size,and non-bonding interactions will be ignored.Therefore,in order to avoid artificial edge effects,the geometry of the outer part of the cluster model was frozen at the original structure and only the innermost part representing the adsorption site of the cluster model was fully optimized.
A lot of researches have been implemented about adsorption and desorption of benzene and its derivatives on clay materials.13-22For example,Castro et al.13,14employed the(Al2Si2O9H4)3cluster model to study the properties(molecular orbital,electrostatic potential,vibrational frequencies,electron localization function(ELF),atomic charges,molecular orbital information,and so on)of benzene interaction with kaolinite surfaces by semi-empirical AM1,RHF,B3LYP,and MP2 methods,and found that the benzene molecule is largely tilted related to the Al―O surface.Michalková et al.18used the Si13O15H35and All6O24H30cluster models to study the adsorption of 2,4-dinitroluene(DNT)on the tetrahedral and octahedral surfaces of dickite at the HF/3-21G level,and they discovered that the adsorption energy of DNT-octahedral fragment system was lower than that of the DNT-tetrahedral fragment.Furthermore,the interaction of 1,3,5-trinitrobenzene(TNB)with the basal siloxane surface of clay minerals was studied by Pelmenschikov and Leszczynski.19The silicon-oxygen clusters symbolizing the siloxane sites were constructed by SiO4tetrahedra with the Si―O bond length equal to 0.161 nm.Gorb et al.20also used Si13O37H22cluster to study the TNB interaction with a pure silicon-oxygen surface.Their studies both suggested that the interaction of TNB with the basal siloxane surface is governed by the balance between favorable dispersion,electrostatic forces,and repulsive exchange forces.Therefore,all the above ab initio results provide reliable references for us to construct a new kaolinite cluster model.
The primary aim of this work is to construct a new cluster model of kaolinite,and to make an extensive investigation on the new cluster model.The various gas state properties characterizing the interaction of benzene molecule and cluster model of kaolinite surface include optimized structures,structural parameters,adsorption energies,secondary hydrogen bonds,electron density characteristics,vibration frequencies,natural bond orbital(NBO)charge distributions,electrostatic potential,and electron density difference,and so on.Simultaneously,the connection among the various properties is indicated.
2 Models and methodology
All quantum chemical calculations were performed at the MP2/6-31G(d,p)//B3LYP/6-31G(d,p)level using the Gaussian 03 suit of programs.23This approach has been shown to be suitable for describing adsorption on oxide surface.13Among clays,kaolinite,composed of one Si―O layer linked to an Al―O layer,is classified as 1:1 type layer aluminosilicate.14,24,25Inspired by Michalková′s work,5we constructed Si6O18H12and Al6O24H30cluster models to represent the Si―O and Al―O layers of kaolinite,respectively(Fig.1).In the center of the Al―O surface,the six O atoms make up a triangle(Fig.2).The Si―O layer consists of SiO4tetrahedrally coordinated Si,and six O atoms of the center form a hexagon(Figs.1-2).It is universally accepted that the“dangling”valences of the border oxygen atoms of the Si―O layer and Al―O layer clusters were saturated with hydrogen atoms.For the optimized structures,all vibrational frequencies were performed at the same basis set level,and the scaled factor of frequencies is 0.9611.26The adsorption energy(AE)for each complex system was calculated as the difference between the total energy of the whole system and the total sum of energies of both subsystems,and the counterpoise corrections for the basis set superposition error(BSSE)were included.

Fig.1 Kaolinite cluster models Si6O18H12(a)and Al6O24H30(b)for Si―O surface and Al―O surface,respectively
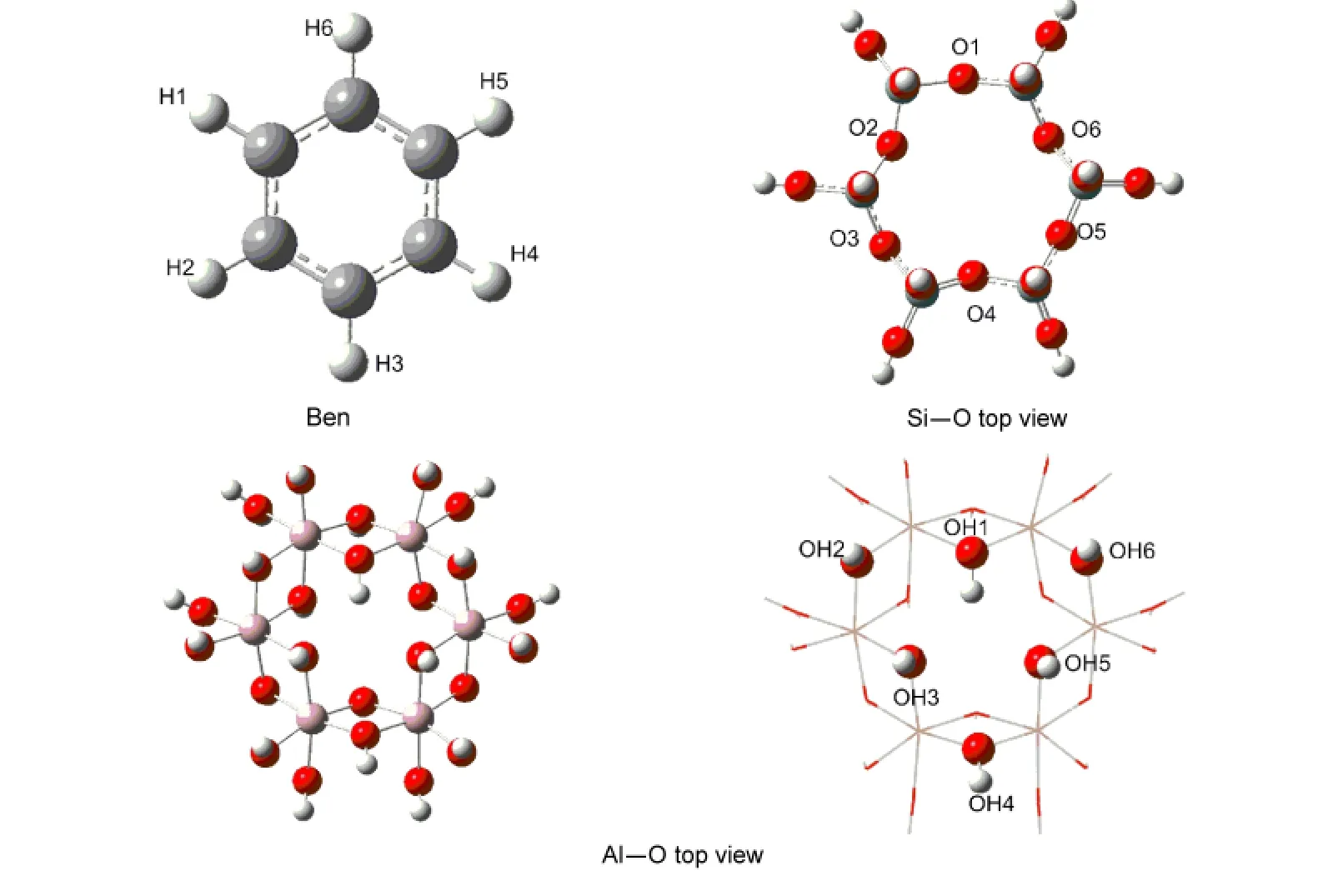
Fig.2 Atomic labels of benzene molecule(Ben)and the innermost part representing the adsorption site of kaolinite cluster model
In this paper,the weak hydrogen bonds C―H…O with 0.23 nm<R(H…O)<0.32 nm,0.32 nm<R(C…O)<0.40 nm and 105°<∠C―H…O<170°are considered to be secondary hydrogen bonds.To gauge the bonding properties,27,28the AIM methodology29was applied to analyze the electron denstiy ρ(r)and its Laplacian(▽2ρ(r))at the critical point of Hδ+…Oδ-from the optimized results at the B3LYP/6-31G(d,p)level.In general,a small electron denstiy ρ(r)at the hydrogen bond critical point(BCP)indicates a weak hydrogen bond,and a positive value of Laplacians ▽2ρ(r)implies that the closed-shell(electrostatic)interaction is the major source in the hydrogen bonded system.Simultaneously,Molekel program package30was applied to obtain the electrostatic potential.The maps of electron density of adsorbed complex system were also studied by the program Multiwfn31and gsgrid.32
3 Results and discussion
3.1 Verification of kaolinite cluster model
To further verify the reasonableness of the kaolinite cluster model,the average bond lengths of kaolinite cluster model have been compared with those from other methods25,33-35(Table 1).The results show that our data are consistent with the others.Besides,interaction energies per water of the intercalation of n(n=1-3)water molecules in kaolinite(K-n W(n=1-3))were examined(Table 2).At the same time,the optimized structures of the intercalation of n(n=1-3)water molecules in kaolinite are depicted in Fig.3.The other detailed information concerning kaolinite-water systems can be found in reference.33We found that the interaction energies per water of kaolinitewater systems are close to those calculated by other density functional theory(DFT)methods.For example,the range of interaction energy per water calculated by Hu and Michaelides34is-43.44--63.70 kJ·mol-1,and our value is-36.69--64.68 kJ·mol-1.Specifically,when the numbers of intercalated water molecule are 2 and 3,the interaction energies per water are-61.78 and-64.68 kJ·mol-1,respectively.These data further confirm the reasonableness of our cluster models.
3.2 Structural and energetic properties
The benzene molecule was respectively optimized on the Si―O and Al―O surfaces of kaolinite at the B3LYP/6-31G(d,p)level.It is worth mentioning that although its initial geometry is parallel to the surfaces,the optimized benzene molecule is almost perpendicular to the surfaces,which is in agreement with Castro′s earlier work,14but different from Castro′s later job.13Various optimized low-energy structures of adsorption ofbenzene molecule on the Al―O surface and Si―O surface of kaolinite are respectively depicted in Figs.4-5.From Figs.4-5,it can be seen that the interaction angle between the benzene ring plane and kaolinite surface for benzene/kaolinite complex is almost close to 90°.In each complex,two H atoms of benzene molecule are preferentially located above the Al―O triangle or Si―O hexagon area,and interact with the surface O atoms by forming C―H…O secondary hydrogen bond.Therefore,the nature of the adsorption of benzene molecule on the kaolinite surfaces may be the formation of secondary hydrogen bonds.The relevant adsorption energies(AE),structural parameters,and electron density characteristics of secondary hydrogen bonds for these low-energy structures are indicated in Tables 3-4.In Table 3,the average values of R(C…O)and R(H…O)are respectively 0.3574 and 0.2498 nm for the lowest energy structure Al-O-Ben1 of benzene adsorption on Al―O surface of kaolinite,less than 0.3583 and 0.2816 nm for the lowest energy structure Si-O-Ben1 of benzene adsorption on Si―O surface of kaolinite.The average values of R(O…H)and∠C―H…O for all of the benzene/Al―O surface complexes are about 0.2630 nm and 143.9°,whereas those for benzene/Si―O surface complexes are about 0.2788 nm and 141.5°,respectivtly.
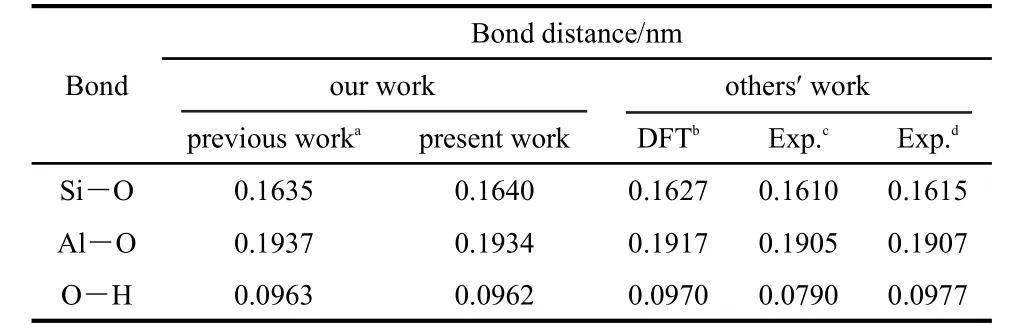
Table 1 Comparison of some typical bond distances of kaolinite cluster model
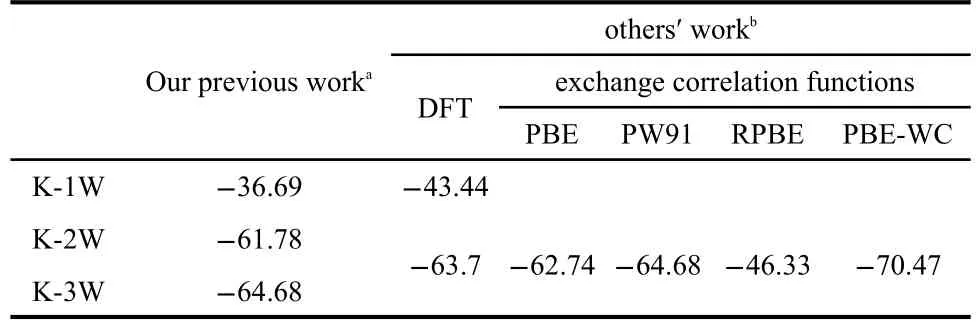
Table 2 Comparison of the interaction energies per water(kJ·mol-1)of the intercalation of n water molecules in kaolinite(K-n W(n=1-3))

Fig.3 Optimized structures of kaolinite-water clusters(K-n W(n=1-3))
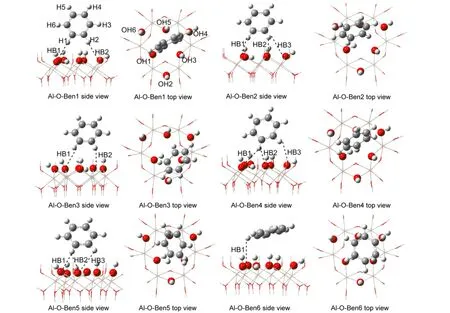
Fig.4 Various low energy structures of adsorption of benzene on Al―O surface of kaolinite
In addition,the AE and conventional counterpoise(CP)method corrected adsorption energies(AECP)between benzene molecule and kaolinite surface have been calculated(Table 4).For Al-O-BEN1,the AECPvalue is-18.40 kJ·mol-1.For Si-OBEN1,the AECPvalue is-18.57 kJ·mol-1.Previous studies36,37have suggested that,for systems with weak interactions,the real BSSE is small.That is to say,the real BSSE can be considered to be one order of magnitude lower than the associated counterpoise correction(BSSE=CP/10).37In this paper,we applied the approximation of BSSE=CP/10.The adsorption energies with real BSSE correction(AEBSSE)for Al-O-Ben1 and Si-O-Ben1 complexes would be-40.85 and-34.04 kJ·mol-1,respectively.This energy difference may be caused by two factors.The first one is the repulsion between π orbital of benzene and the orbitals of oxygen network from the kaolinite Si―O surface.The second one is the size of energy gap between HOMO(the highest occupied molecular orbital)and LUMO(the lowest unoccupied molecular orbital)for different clay surface.The greater the energy gap,the lower the surface chemical activity,the more stable the surfaces.Si―O surface has greater energy gap(6.52 eV)than Al―O surface(4.20 eV).This further shows that the Si―O surface is more stable than the Al―O surface.As a final result of the above two factors,the benzene molecule will be adsorbed more preferably on the Al―O surface than on the Si―O surface.
In the current investigation,based on the analysis of electron density characteristics(Table 3),we confirmed the existence of secondary hydrogen bonds for structures in Figs.4-5.The results show that the ρ and ▽2ρ values of secondary hydrogen bonds for Al-O-Ben complexes are respectively 0.00364-0.01361 a.u.and 0.01384-0.03704 a.u.except for the HB1 and HB2 of Al-O-Ben3.However,the secondary hydrogen bonds of Si-O-Ben complexes,which are characterized by 0.00197-0.00846 a.u.of ρ values and 0.00894-0.02735 a.u.of▽2ρ values,are slightly weaker than those of Al-O-Ben complexes.At the same time,the values of the electron density and the Laplacian of the electron density are proportional to the strength of formed hydrogen bonds(shorter distances corresponding with larger ρ and ▽2ρ values).
3.3 NBO charge distributions
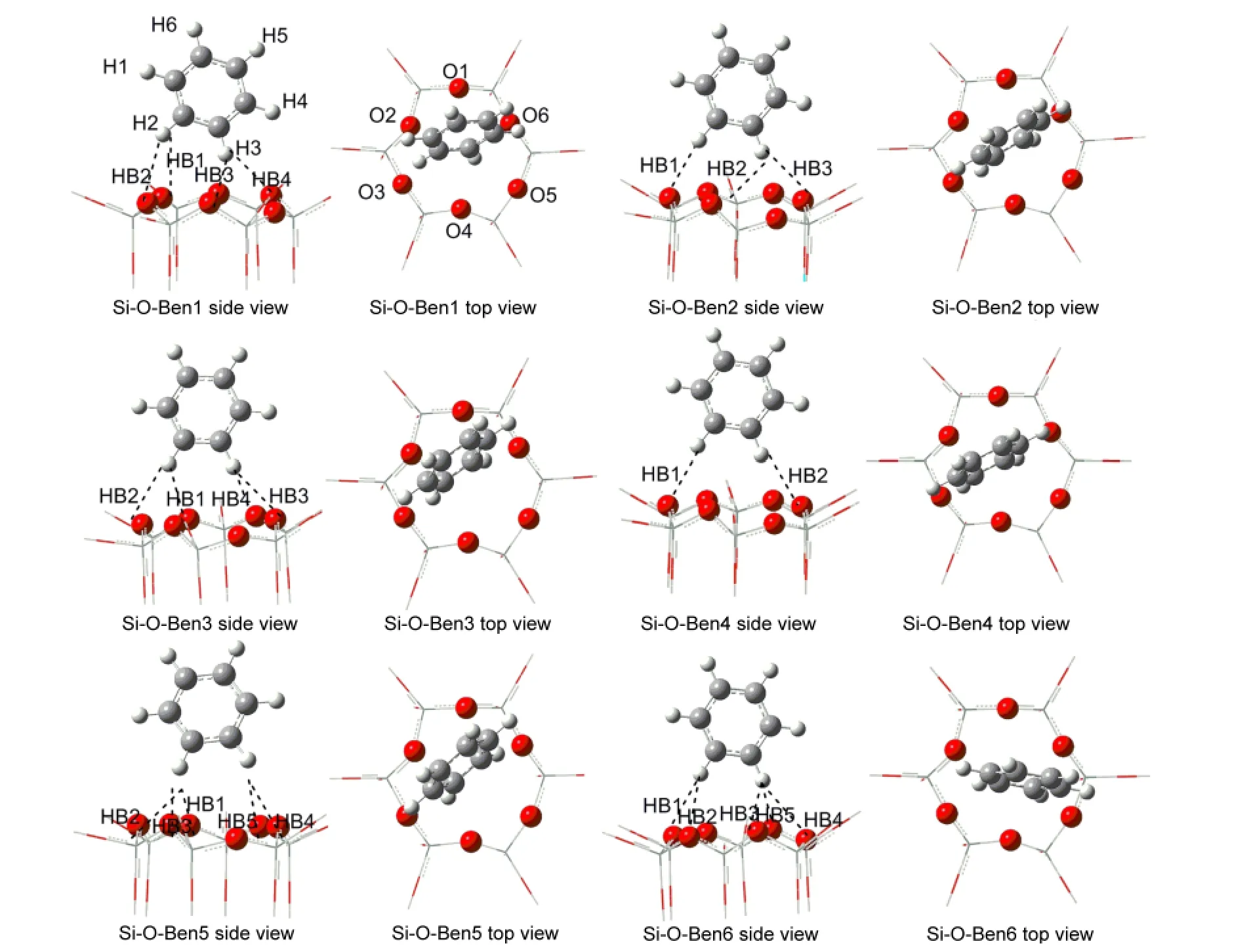
Fig.5 Various low energy structures of adsorption of benzene on Si―O surface of kaolinite

Table 3 Structural parameters for benzene/kaolinite adsorbed systems and electron density characteristics(ρ,▽2ρ)of hydrogen bonds both calculated at the MP2/6-31G(d,p)//B3LYP/6-31G(d,p)level

Table 4 AE,CP method and real BSSE corrected adsorption energies(AECPand AEBSSE)for benzene/kaolinite adsorbed systems calculated at the MP2/6-31G(d,p)//B3LYP/6-31G(d,p)level
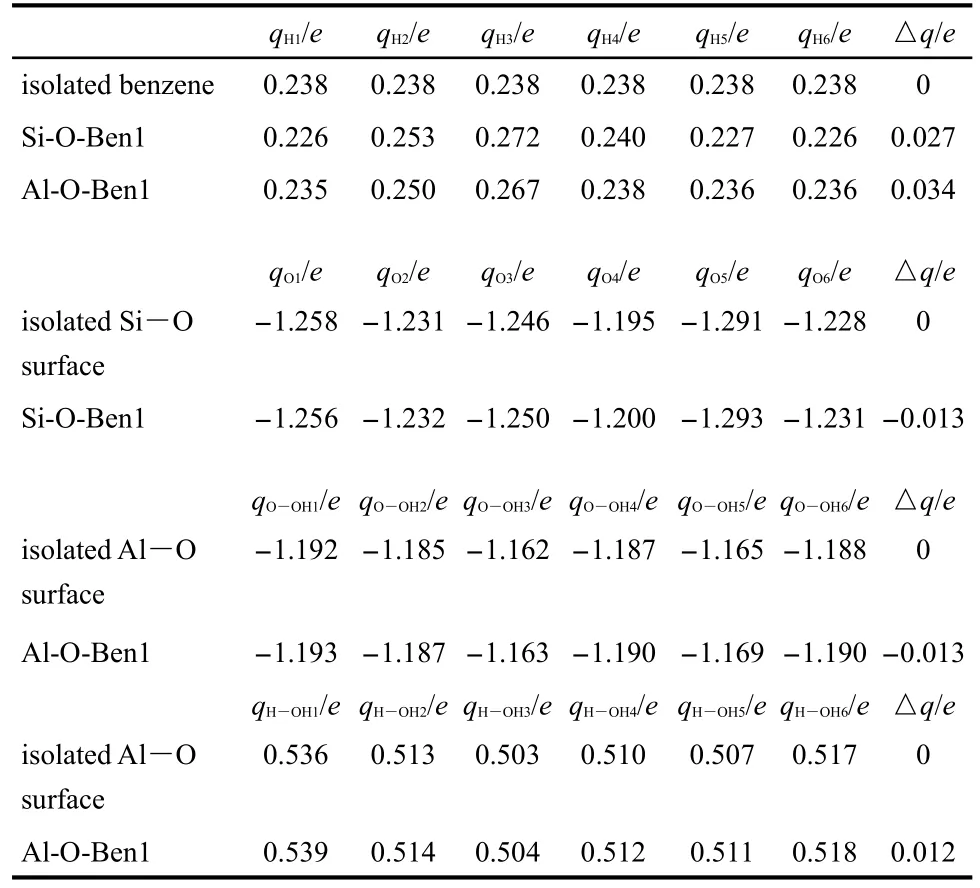
Table 5 NBO charges of the lowest-energetic structure for benzene and kaolinite surfaces calculated by the B3LYP/6-31G(d,p)method
In this section,a cluster structure is arbitrarily chosen to study the NBO charge distributions.Taking the lowest energy structures Al-O-Ben1 and Si-O-Ben1 for example,the charges are listed in Table 5.For the isolated benzene molecule,the charge change of hydrogen atoms is regarded as zero.When benzene molecule interacts with each kaolinite surface,the charges of all sites for benzene molecule are different from the isolated benzene.i.e.,for the isolated benzene,no other molecule affects its electron cloud,but for a benzene/kaolinite complex,the intermolecular secondary hydrogen bonds C―H…O directly influence the charge distribution.For example,for the structure Si-O-Ben1,two H atoms(qH2and qH3are 0.253e and 0.272e)exhibit relatively larger absolute charges than the corresponding H atoms(0.238e and 0.238e)of isolated benzene,because the two H atoms and oxygen atoms of Si―O surface form the hydrogen bonds.Simultaneously,the charge change(△q)of all H atoms is 0.027e.For the structure Al-O-Ben1,△q of all H atoms is 0.034e,which is slightly larger than 0.027e of Si-O-Ben1.Thus,it indicates that the polarization degree of benzene on Al―O surface is slightly greater than that on Si―O surface.
In the same way,this is also true of Al―O and Si―O surfaces.From Table 5,we can also see that after benzene adsorption on the Si―O surface,the absolute charges of O atoms of Si―O surface are almost all a little bit increased.Specifically,for the structure Si-O-Ben1,│Δq│of O atoms of Si―O surface is 0.013e.The same results occurred on the Al―O surface.H atoms and O atoms of Al―O surface for structure Al-O-Ben1 both exhibit relatively larger absolute charge(0.012e and 0.013e)than those of the isolated Al―O surface.
To sum up,we hold that the polarization degrees of O atoms of Al―O surface are stronger than that of the Si―O layer,and further validate that benzene molecule is adsorbed more preferably on the Al―O surface than on the Si―O surface.
3.4 Vibration frequencies
We have also calculated the vibration frequencies of benzene as well as the lowest energy structures for the benzene adsorption on Al―O/Si―O surface by the B3LYP/6-31G(d,p)method and the related data are shown in Table 6.After benzene was adsorbed on the Al―O surface and Si―O surface,bending vibration frequencies for plane and out of plane have been enhanced.For examples,C―H bending vibration frequencies for plane of Si-O-Ben1 and Al-O-Ben1 complexes are respectively 1132-1159 cm-1and 1137-1170 cm-1,slightly larger than 1133-1155 cm-1of the isolated benzene.At the same time,C―H bending vibration frequencies for out of plane also increase from 830-973 cm-1of the isolated benzene to 834-991 cm-1of Al-O-Ben1 and 840-973 cm-1of Si-OBen1.Nevertheless,Table 6 indicates a little decrease of C―H and C=C stretching vibrations of adsorbed benzene,comparedto free benzene molecule.Specifically,C―H stretching vibration frequencies of Si-O-Ben1 and Al-O-Ben1 are respectively 3032-3080 cm-1and 3044-3076 cm-1,lower than 3051-3086 cm-1of the isolated benzene,and C=C stretching vibration frequencies also decrease from 1302-1589 cm-1of the isolated benzene to 1301-1585 cm-1of Si-O-Ben1 and 1302-1585 cm-1of Al-O-Ben1.Furthermore,O―H stretching vibration frequencies of Al―O surface change a little bit,which decrease from 3582-3808 cm-1(from OH1 to OH6)of the isolated Al―O surface to 3567-3802 cm-1of Al-O-Ben1.These results are in accordance with the Castro′s earlier work14(for details see Table 6).To a lesser extent,these changes of vibration frequencies,especially the stretching modes of hydroxyl groups of kaolinite surface and C―H of benzene ring,can account for the adsorption between benzene and kaolinite surface.So people can utilize kaolinite to remove organics and remediate soils and groundwater contaminated with petroleum hydrocarbons.
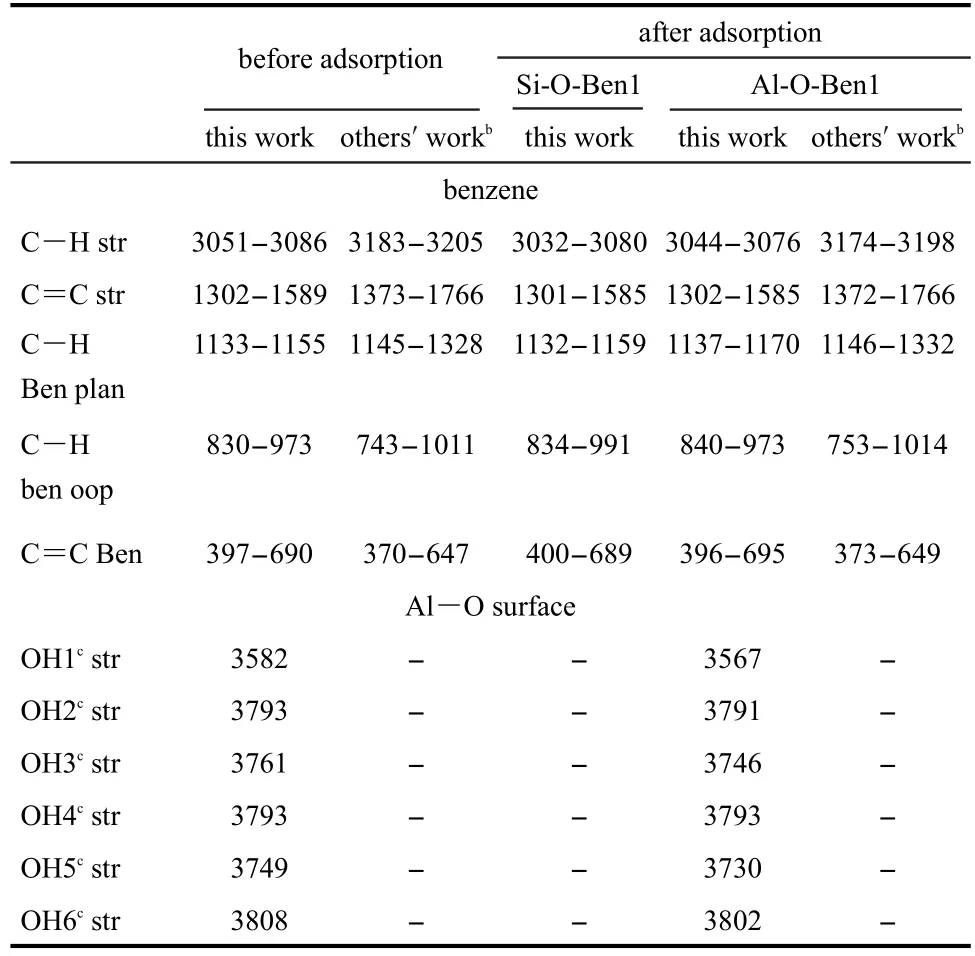
Table 6 Comparison of vibration frequenciesa(cm-1)of benzene and benzene/kaolinite complex Si-O-Ben1 and Al-O-Ben1
3.5 Electrostatic potential on adsorption
The isopotential electrostatic surfaces of the lowest energy structures are depicted on Fig.6 for benzene,kaolinite,and benzene/kaolinite complex.It is universally acknowledged that for adsorption of organic molecules,the calculation of maps of electrostatic potentials(MEPs)on the surface of a solid material can help us acquire much more knowledge about the adsorption processes.For adsorption of the benzene ring on clay minerals,in which weak interactions are originated from hydrogen bonds,it is interesting to calculate the electrostatic potential between the adsorbate and substrate because it helps to determine the related properties of sorption complexes.The electrostatic potential of benzene adsorbed on kaolinite surfaces showed no obvious difference compared to the isolated kaolinite surface.The results of electrostatic potential show the same tendency for the interaction of benzene molecule with the Al―O and Si―O surfaces.Furthermore,the adsorption of benzene ring on the Al―O surface results in much less significant changes in the MEPs than that on the Si―O surface(Fig.6).It indicates that the formation of the secondary hydrogen bonds causes much less significant polarization of the target molecule,because Δq of all H atoms of benzene molecule are only+0.034e and+0.027e(Table 4)for Al―O and Si―O surfaces,respectively.
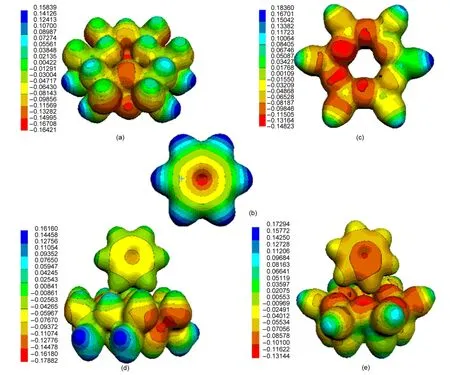
Fig.6 Surfaces of electrostatic potentials(unit in a.u.)for(a)Al―O surface cluster modelAl6O24H30,(b)benzene molecule,(c)Si―O surface cluster model Si6O18H12,the lowest energy structures(d)Al-O-Ben1 and(e)Si-O-Ben1 of benzene/kaolinite complex

Fig.7 (a)Surfaces of electron density difference for the lowest-energy structure Al-O-Ben1 of benzene/Al―O surface complex;(b)enlargement of the left side of the hydrogen bonds;(c)enlargement of the right side of the hydrogen bonds
3.6 Electron density difference
An examination of Fig.7 indicates that the common trend is that electron density decreases on carbon atoms of benzene molecule and oxygen atoms of Al―O surface,while electron density increases on the basis of the C―H bond and C=C bond.In the interaction region of secondary hydrogen bond C―H…O,electrons on the oxygen atoms of Al―O surface are gathered to the middle of benzene molecule and Al―O surface,therefore electron densities of oxygen atoms on the Al―O surface apparently diminish.On the contrary,the electron densities between benzene molecule and Al―O layer increase simultaneously.However,the electron densities of O atoms away from the H atoms in the benzene ring increase a little bit,as we can see clearly from Fig.7(b),which is consistent with the increase of NBO charge(Table 5)absolute value of oxygen atoms on the Al―O surface.Additionally,with the decrease of hydrogen atom electron densities around,the NBO charges(Table 5)of the corresponding hydrogen atoms increase.Nonetheless,these small changes will have a positive effect on the energy of adsorption,so that,there is,to a small extent,a chemical contribution to the adsorption mechanism.
The change of electron density suggests that enhanced polarization leads to a predominant electrostatic interaction between benzene molecule and kaolinite surface.It also indicates that the adsorption results in significant electron transfer between the molecule and the surface.We also can draw the same conclusion from the electron density characteristics of Table 5.Additionally,the electron density is transferred to the π orbital of benzene and comes mainly from the C―H…O hydrogen bonding,depicted clearly in Fig.7.All these can be accounted for the gradual formations of C―H…O secondary hydrogen bonds.
4 Conclusions
The interactions of benzene with the kaolinite cluster models,Si6O18H12(Si―O layer)and Al6O24H30(Al―O layer),have been systematically investigated at the MP2/6-31G(d,p)//B3LYP/6-31G(d,p)level to mimic the adsorption of benzene on the kaolinite surface.Various gas state properties characterizing benzene/kaolinite complexes,including the optimized structures,structural parameters,adsorption energies,NBO charge distributions,vibration frequencies,and electrostatic potential,electron density characteristics,and electron density difference,were presented and discussed in this paper.The optimized structures show that there are secondary hydrogen bond interactions between the benzene molecule and the polar kaolinite surface.That is to say,the nature of the adsorption of the benzene molecule on kaolinite surfaces may be the formation of secondary hydrogen bonds.The results of other properties further confirmed the existence of secondary hydrogen bonds.Simultaneously,benzene molecule is more likely to be adsorbed on the Al―O surface than on the Si―O surface,and the absorption angle between benzene ring plane and kaolinite Al―O surface is about 90°.
(1)Roldan,R.;Romero,F.J.;Jimenez,C.;Borau,V.;Marinas,J.M.Appl.Catal.A 2004,266,203.doi:10.1016/j.apcata.2004.02.008
(2)Maliyekkal,S.M.;Rene,E.R.;Philip,L.;Swaminathan,T.J.Hazard.Mater.2004,109,201.doi:10.1016/j.jhazmat.2004.04.001
(3)Pilidis,G.A.;Karakitsios,S.P.;Kassomenos,P.A.;Kazos,E.A.;Stalikas,C.D.Environ.Monit.Assess.2009,150,285.doi:10.1007/s10661-008-0230-9
(4)Lesage,S.Anal.Chem.ACS Publications 1993,65,647A.
(5)Michalková,A.;Tunega,D.;Nagy,L.T.J.Mol.Struct.-Theochem 2002,581,37.doi:10.1016/S0166-1280(01)00741-2
(6)Hess,A.C.;Saunders,V.R.J.Phys.Chem.1992,96,4367.doi:10.1021/j100190a047
(7)Shua,H.T.;Lib,D.;Scalaa,A.A.;Mab,Y.H.Purif.Technol.1997,11,27.doi:10.1016/S1383-5866(96)01005-2
(8)Beltrán,A.;Andrés,J.;Calatayud,M.;Martins,J.B.L.Chem.Phys.Lett.2001,338,224.doi:10.1016/S0009-2614(01)00238-X
(9)Martins,J.B.L.;Sambrano,J.R.;Vasconcellos,L.A.S.;Longo,E.;Taft,C.A.Quim.Nova.2004,27,10.doi:10.1590/S0100-40422004000100003
(10)Martins,J.;Taft,C.;Lie,S.;Longo,E.J.Mol.Struct.-Theochem 2000,528,161.doi:10.1016/S0166-1280(99)00498-4
(11)Almeida,A.;Martins,J.;Longo,E.;Taft,C.;Murgich,J.;Jalbout,A.F.J.Mol.Struct.-Theochem 2003,664,111.
(12)Sambrano,J.;Vasconcellos,L.;Martins,J.;Santos,M.;Longo,E.;Beltran,A.J.Mol.Struct.-Theochem 2003,629,307.doi:10.1016/S0166-1280(03)00200-8
(13)Castro,E.A.S.;Gargano,R.;Martins,J.B.L.Int.J.Quantum Chem.2012,112,2828.doi:10.1002/qua.v112.16
(14)Castro,E.A.S.;Martins,J.B.L.J.Comput.Aided Mater.Des.2005,12,121.
(15)Bickmore,B.R.;Rosso,K.M.;Nagy,K.L.;Cygan,R.T.;Tadanier,C.J.Clays Clay Min.2003,51,359.
(16)Castro,E.A.S.;Martins,J.B.L.Int.J.Quantum Chem.2005,103,550.
(17)Balan,E.;Saitta,A.M.;Mauri,F.;Lemaire,C.;Guyot,F.Am.Mineral.2002,87,1286.
(18)Michalková,A.;Szymczak,J.J.;Leszczynski,J.Struct.Chem.2005,16,325.doi:10.1007/s11224-005-4463-8
(19)Pelmenschikov,A.;Leszczynski,J.J.Phys.Chem.B 1999,103,6886.doi:10.1021/jp990091q
(20)Gorb,L.;Lutchyn,R.;Zub,Y.;Leszczynska,D.;Leszczynski,J.J.Mol.Struct.-Theochem 2006,766,151.doi:10.1016/j.theochem.2006.04.013
(21)Lee,J.F.;Mortland,M.M.;Chiou,C.T.;Kile,D.E.;Boyd,S.A.Clay Clay Min.1990,38,113.
(22)Wilson,M.A.;Lee,G.S.H.;Taylor,R.C.Clay Clay Min.2002,50,348.
(23)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,Revision A.01;Gaussian Inc.:Pittsburgh,PA,2003.
(24)Young,R.A.;Hewat,A.W.Clay Clay Min.1988,36,225.
(25)Bish,D.L.Clay Clay Min.1993,41,738.
(26)Karl,K.I.;Russell,D.J.;Raghu,N.K.J.Phys.Chem.A 2005,109,8430.doi:10.1021/jp052793n
(27)Koch,U.;Popelier,P.J.Phys.Chem.1995,99,9747.doi:10.1021/j100024a016
(29)Bader,R.W.F.Accounts Chem.Res.1985,18,9.
(30)Flükiger,P.;Lüthi,H.;Portmann,S.;Weber,J.Molekel 4.0;Swiss Center for Scientific Computing:Manno,Switzerlan,2000.
(31)Lu,T.;Chen,F.J.Comput.Chem.2012,33,580.doi:10.1002/jcc.v33.5
(32)Lu,T.GsGrid:Extracting Data from Gaussian Grid File and Grid File Calculation[EB/OL].http:∥gsgrid.codeplex.com,in.
(33)Zhang,C.;Song,K.H.;Wang,X.;Yin,H.Z.;Qian,P.J.Mol.Sci.2013,29,134.[张 超,宋开慧,王 幸,尹洪宗,钱 萍.分子科学学报,2013,29,134.]
(34)Hu,X.L.;Michaelides,A.Surf.Sci.2008,602,960.doi:10.1016/j.susc.2007.12.032
(35)Neder,R.;Burghammer,M.;Grasl,T.;Schulz,H.;Bram,A.;Fiedler,S.Clay Clay Min.1999,47,487.
(36)Austen,K.F.;White,T.O.H.;Marmier,A.;Parker,S.C.;Artacho,E.;Dove,M.T.J.Phys:Condes.Matter 2008,20,035215.doi:10.1088/0953-8984/20/03/035215
(37)Sainz-Díaz,I.;Francisco-Márquez,M.;Vivier-Bunge,A.Environ.Chem.2011,8,429.
