空气(氧气)催化氧化合成草甘膦催化剂的研究进展
2014-10-13陈丹李国儒黄敏建陈标华银凤翔1
陈丹 ,李国儒,黄敏建,陈标华,银凤翔1,
(1北京化工大学有机无机复合材料国家重点实验室,北京 100029; 2北京化工大学化学工程学院,北京 100029;3北京化工大学常州先进材料研究院,江苏 常州 213164)
草甘膦,化学名称N-磷酰基甲基甘氨酸,通用名称glyphosate,分子式C3H8NO5P,是一种低毒、高效、广谱且对环境友好的灭生性除草剂[1]。
自20世纪70年代美国孟山都公司开发成功后,全球对草甘膦的需求迅速增长,大批草甘膦生产企业如雨后春笋般涌现,导致了草甘膦市场趋于饱和,企业的利润空间缩小,企业竞争加剧[2]。为了提高竞争力,企业不得不在降低生产成本的同时提高产品质量,所以草甘膦生产工艺的选择和原料的使用尤为重要。
目前国际上草甘膦的主流合成工艺是氢氰酸-亚氨基二乙酸(IDA)路线,这种路线首先以氢氰酸为原料合成中间体亚氨基二乙酸,然后亚氨基二乙酸再与亚磷酸、甲醛等反应合成双甘膦(PMIDA),双甘膦再经催化氧化反应最终转化为草甘膦(PMG)。其中,双甘膦催化氧化合成草甘膦是该工艺路线的关键步骤,此过程中双甘膦的转化率、草甘膦的收率以及纯度决定了整个工艺路线的生产成本和产品质量,其反应方程式如式(1)。
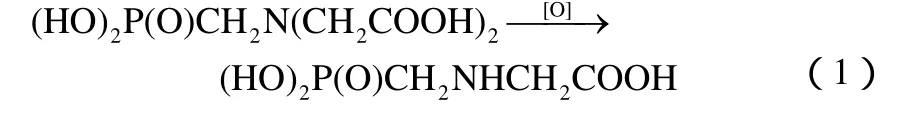
双甘膦催化氧化合成草甘膦的核心问题是氧化剂、氧化方法和催化剂的选择。高效的催化剂、合适的氧化剂及氧化方法对于草甘膦的收率和整个工艺路线的经济效益都有关键影响[3-4]。以空气或氧气作为氧化剂的催化氧化双甘膦合成草甘膦的方法属于绿色化学的范畴,具有工艺路线简单、产品质量好、环境友好等优点,是目前双甘膦氧化合成草甘膦最理想的方法,因此最具研究价值。该方法的核心是高效催化剂的选择,催化剂不但能够降低反应活化能、加快反应速率,还可以提高反应物的转化率和产物收率,对整个工艺过程起着至关重要的作用,因此,近年围绕研发高效催化剂进行了大量的研究工作,也取得了一系列研究成果。从催化剂的研究情况来看,主要集中在对活性炭催化剂、贵金属催化剂和过渡金属催化剂的研究开发上,因此,本文主要对这些催化剂近年的研发进展进行综述。
1 活性炭催化剂
活性炭不但可以作为催化剂的载体,而且本身也可以作为催化剂使用[5]。通过适当的热处理或化学处理,可以在活性炭表面引入含杂原子(N、O等)的官能团,这些官能团都是潜在的活性位点,正是由于这些活性位点的引入使得活性炭具有催化作用,并广泛用于选择性催化氧化、氯化和脱氯等反应。采用活性炭作催化剂催化氧化合成草甘膦不仅成本低,反应的收率几乎可达 100%,而且可以得到纯度很高的草甘膦。1976年,美国孟山都公司的 Hershman[6]首先发明了用活性炭作催化剂合成草甘膦的方法,开创了活性炭催化氧化双甘膦合成草甘膦的先例。经过详细的实验研究,Hershman确定了活性炭催化氧化反应的最佳条件为:使用比表面积为400~1600 m2/g、粒度<0.044 mm的活性炭作催化剂,含分子氧气体作氧化剂,在反应压力为0.1~0.7 MPa、反应温度为 75~150 ℃的条件下,草甘膦的收率可达100%,但是产品质量不够稳定。国内的韦少平等[7]使用比表面积≥1000 m2/g的工业级活性炭作催化剂,分别考察了反应物质量分数、反应温度、反应压力、反应时间等因素对草甘膦收率的影响,并优化了反应条件,在优化条件下草甘膦的收率可达96%。这种方法虽然以较低的成本得到了较高收率的草甘膦,但缺点是双甘膦初始浓度较低,无形中降低了生产效率。为了克服这一缺点,科研人员进行了大量的研究工作。周曙光等[8]以煤质和椰壳炭化料为原料,使用传统的水蒸气活化法制得了微孔结构发达的煤基和椰壳基活性炭催化剂,并使用NH3高温焙烧的方法对催化剂进行了表面还原改性,改性后催化剂的微孔孔容、比表面积和N元素含量都明显增加了。以高纯氧气作氧化剂,使用改性后的催化剂,对高浓度双甘膦间歇氧化合成草甘膦工艺进行了优化,在双甘膦初始浓度为40 %(质量分数),m(催化剂)∶m(双甘膦)= 0.189、反应温度为45℃、反应压力为0.5 MPa、反应时间为 5 h的条件下,草甘膦的总收率为 97.1%,纯度>97.0%。
除了进行工艺条件优化、开发新的活性炭催化剂之外,活性炭催化氧化合成草甘膦的反应机理也得到了系统深入的研究,为合成高效催化剂提供了理论依据,其催化氧化机理如图1所示。
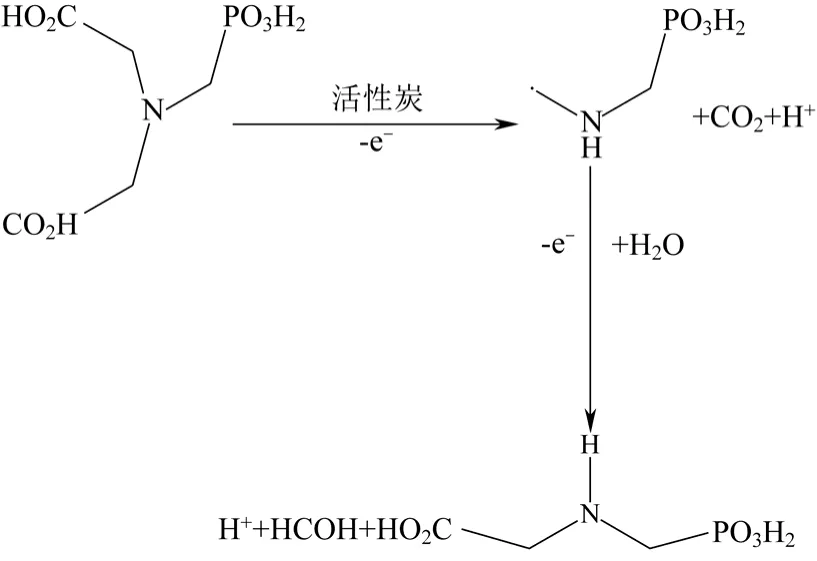
图1 活性炭催化氧化合成草甘膦机理[9]
活性炭催化氧化双甘膦合成草甘膦的反应是一个氧化脱羧的过程,活性炭可以促进双甘膦的断裂,而起脱羧作用的主要是活性炭表面上的官能团。Besson等[10]考察了不同来源和特征的活性炭催化剂对双甘膦催化氧化合成草甘膦反应的影响,结果表明,比表面积小的活性炭的比活性较差,然而面积速率的比较表明活性与比表面积没有直接关系。深入研究发现这可能是活性炭表面的氧化性官能团的影响。活性炭表面的酸性基团对催化活性有负面影响,可以通过热处理的方法减少活性炭表面酸性基团的数量,处理温度低于700 ℃对活性无影响,而900 ℃热处理可以使催化活性提高40%,这是因为高温热处理生成了对反应有利的含氮基团[11]。Chou[12-13]也提出活性炭表面的酸性基团会降低活性炭的催化活性,除去活性炭表面的酸性基团可以极大地提高其催化活性。另外,Pinel等[5]的研究也表明活性炭表面含有含氮官能团,这种官能团一方面由活性炭制备时的原料带入,另一方面是在改性处理过程中生成。活性炭经氮气 900 ℃高温处理后,可以在很大程度上加快反应速率;如果在900 ℃用氨气高温处理活性炭,催化速率则达到最大,这是因为用氮气或氨气热处理减少了活性炭表面酸性位点的数量,增加了对反应有利的碱性活性位点的数量。
2 贵金属催化剂
用于空气(氧气)催化氧化合成草甘膦的贵金属催化剂一般是活性炭负载的贵金属催化剂,其中活性炭的作用是促进双甘膦氧化脱羧生成草甘膦和甲醛,而贵金属则主要使甲醛氧化成甲酸,甲酸进一步氧化转化为CO2和水。贵金属的使用是很有必要的:①副产物甲醛对环境有危害,高含量甲醛污水的处理增加了生产成本,所以为了降低副产物甲醛的含量就需要使用贵金属催化剂,另外,如图 2所示,副产物甲醛和甲酸等会与主产物 PMG发生连串反应,生成副产物N-methyl-PMG等,从而导致 PMG的收率降低;②负载贵金属的活性炭可以多次套用,而活性仍然稳定,反而没有负载贵金属的活性炭催化剂每套用一次催化活性就要下降约10%[14]。
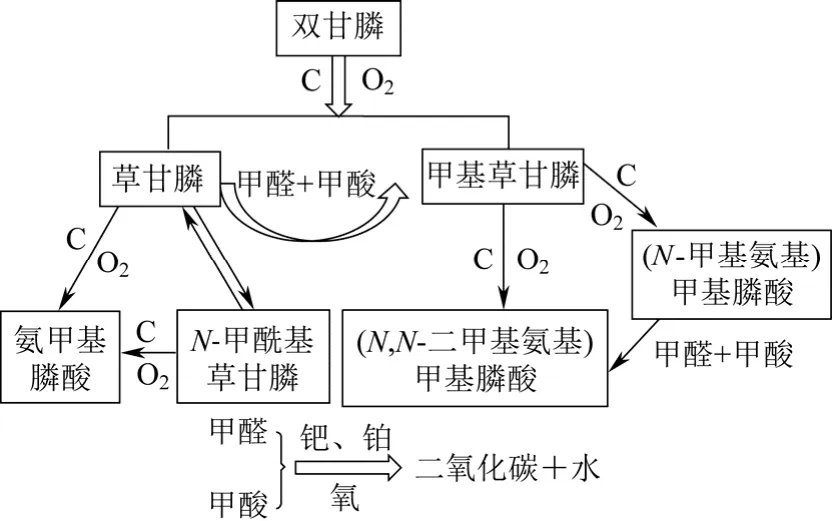
图2 双甘膦氧化产物转化关系图[4]
贵金属催化剂催化氧化双甘膦合成草甘膦不但选择性好、收率高,而且得到的草甘膦质量也很好。常用于负载的贵金属有钯、铂、铑、钌、银、铱、金等。孟山都的 Franze[15]将贵金属 Pd、Pt、Rh等负载到活性炭上,并以氧气为氧化剂,研究了这些催化剂催化氧化合成草甘膦的性能,结果表明,在压力为0.207~7.166 MPa、温度为90~120 ℃、反应时间为1~3 h的条件下,草甘膦的收率为96%,纯度高达97%。国内的陈彰明等[16]使用氯铂酸作前体制备了Pt/AC催化剂,考察了反应温度、反应压力、反应时间、催化剂用量以及原料双甘膦纯度等因素对双甘膦催化氧化合成草甘膦反应的影响。在反应温度为100 ℃、反应压力为0.6 MPa及催化剂用量为6 g/mL时,反应5 h的效果较优,此时双甘膦转化率可达100%,草甘膦的收率为91.9%。刘金红等[17]采用浸渍法将钯和铂负载在活性炭上制备了活性炭负载的贵金属催化剂,考察了载体的不同脱氧预处理方式、贵金属负载方法、贵金属种类及还原方法对催化氧化合成草甘膦性能的影响,结果表明,使用不同处理剂脱氧预处理的载体其催化剂性能有很大差别;负载方法选择干浸法比湿浸法效果更好;铂炭催化剂比钯炭的性能更高;采用气液串联的还原方式得到的催化剂效果最佳,在反应温度为90 ℃和反应压力为0.34 MPa的条件下,使用Pt/AC催化剂合成草甘膦的收率为 75.4%,草甘膦纯度为97.6%。
贵金属催化剂的优良催化效果得到了一致认可,但是这种催化剂的缺点也是不容忽视的:贵金属本身价格昂贵;贵金属很难均匀分散在活性炭上,导致必须要有足够的贵金属负载量才可达到好的催化效果,这极大地增加了催化剂成本;贵金属遇胺类(双甘膦)易中毒失活,被氧化成更稳定的状态,从而与双甘膦和草甘膦结合生成络合物,这些络合物反过来又会溶解贵金属,造成贵金属严重损耗;贵金属容易失活且再生困难。为了克服这些问题,科技工作者进行了大量的研究工作。黄伟等[18]研究了载体预处理、浸渍液、活性组分的还原以及催化剂干燥等因素对活性炭负载 Pd催化剂的分散度以及分布厚度的影响,指出催化剂的制备工艺中吸附液的选择会极大地影响催化剂的活性,吸附液中添加有机溶剂或氧化剂都可以提高钯在活性炭表面的分散度,改善钯在活性炭上的分布厚度。为了克服贵金属易淋失的缺点,可以选择将催化剂做成胶囊的形式或采用母液循环等方法。Felthouse[19]提出了用微晶载体负载贵金属的方法,这种方法实际上是将贵金属做成胶囊的形式,这种特殊的胶囊可以选择性地让甲醛等透过被氧化,而贵金属则留在孔洞中无法透过。但是这种方法还存在一些问题尚未解决,如贵金属的比率难以设计,给催化剂的制备带来了困难,而且难以再生[20]。Hitzler等[21]采用母液循环的方法既减少了贵金属的淋失也提高了草甘膦的收率;Martin Ramon等[22]通过氮气吹洗反应后溶液大大降低了贵金属的流失(从30%降低到1%)。为了防止贵金属催化剂遇到胺类化合物而中毒失活,Hershman等[23]采取在贵金属催化剂表面包裹一层高分子膜的方法,这种膜的特点是选择性透过甲醛、甲酸和氧气等反应物,阻止胺类物质透过,将这种贵金属催化剂应用于催化氧化制备草甘膦的反应中,可以将副产甲醛的含量控制在0.5%以下。为了提高草甘膦的收率,也可以采用添加辅助催化剂的方法,Leiber等[24-26]在Pt/AC或Pd/AC中添加铋(Bi)或碲(Te)等作辅助催化剂,可以提高反应的收率和草甘膦的选择性。目前,美国孟山都公司和陶氏益农公司都采用活性炭负载贵金属作催化剂,空气催化氧化双甘膦的方法来合成草甘膦[27]。国内使用这种方法合成草甘膦目前已经到了中试阶段[4],但降低贵金属催化剂的成本仍是我国草甘膦生产企业亟待解决的问题。
3 过渡金属催化剂
为了克服贵金属催化剂价格昂贵的缺点,科研工作者开发了过渡金属催化剂。由于过渡金属作催化剂无需负载在载体上,而是直接以水溶性盐或络合物的形式加入反应体系中,所以过渡金属催化氧化双甘膦制备草甘膦的反应属于均相催化过程,反应结束后无需过滤催化剂及与草甘膦分离,操作简单,成本也较低。常见的过渡金属如铝、铁、镍、锰、钴、铅、铬、钌、钼、钒、银、锡、铈等过渡金属的络合物或盐都可作为催化氧化制备草甘膦的催化剂。孟山都公司的 Riley等[28-31]对锰、钒、钴等金属盐的催化性能进行了研究,结果表明,在一定的条件下,金属盐的催化活性为:钒最好,锰次之,钴最次;反应的优选温度为70~120 ℃,优选氧气分压为3.1~13.8 MPa。钒的催化条件温和,反应速率较快但是选择性低;锰的催化速率也很快,但是它易与双甘膦形成络合物沉淀 Mn(PMIDA)2;钴的氧化速率不如钒好,但是选择性比钒要好。研究表明[30],钴和锰催化氧化的前体选择乙酸盐较好,在反应温度为70~100 ℃、反应压力为0.207~6.89 MPa、过渡金属离子浓度为 0.1~0.001 mol/L的条件下,催化剂的活性最好。石峰等[32]使用硫酸锰作催化剂,考察了温度、时间、压力和催化剂用量等因素对氧气催化氧化双甘膦合成草甘膦反应收率的影响,在反应温度为 80 ℃、反应时间为 90 min、反应压力为0.2 MPa、催化剂与双甘膦的摩尔比为1∶0.04的优化条件下,得到草甘膦的收率为75.2%,纯度为71.1%。
虽然过渡金属催化剂的成本远远低于贵金属催化剂,但是无论从反应的收率还是除甲醛的效果来看,过渡金属催化剂都与贵金属催化剂存在较大差距。为了进一步提高过渡金属催化剂的催化性能,Fields等[33]采用锰和钴的盐或络合物作为催化剂,在反应体系中加入溴离子,提高了反应的选择性和转化率。溴离子和双甘膦的摩尔比优选0.05~0.4,摩尔比大于0.4或小于0.05都会对反应不利;溴前体选择溴化钠和六水合二溴化钴时催化剂性能最优,此时双甘膦的转化率达96%,草甘膦的选择性达 90.8%;而且反应溶液的酸碱性最好,是自然的未经调节的pH值。钴作催化剂时,双甘膦初始浓度选择过饱和的於浆可以得到最大的收率,但是锰作催化剂时,Mn(Ⅱ)易与 PMIDA形成不溶的Mn(PMIDA)2络合物,为了防止锰沉淀,双甘膦的初始质量分数不得超过 6%。另外,使用钒作催化剂时,加入适当的助催化剂可以提高草甘膦的选择性,比如加入氧化剂醌和双奎尼双吡啶盐可以使草甘膦的选择性提高到96%[34]。
4 结语与展望
空气(氧气)催化氧化法合成草甘膦属于绿色化学范畴,具有工艺路线简单、产品质量好且环境友好的优点,是目前双甘膦氧化合成草甘膦最理想的方法,无论从科技进步还是从工业发展来讲,都极具研究价值。但是这种方法仍然存在着一些缺陷:单独使用活性炭催化剂合成的草甘膦中含有大量的甲醛,难以达到国家标准,而且催化剂套用次数少;贵金属催化剂昂贵的价格令人不得不望而却步;使用过渡金属催化剂,草甘膦的收率和除甲醛效果也不尽人意,而且国内的催化剂制备技术与国外仍存在一定的差距。因此,开发出一种催化效果更优、生产成本更低廉、使用和再生简单且环境友好的催化剂是亟待解决的问题。
[1]陈云. 除草剂草甘膦的性质和应用[J]. 湖北化工,1995(2):10-23.
[2]汪家铭. 草甘膦发展概况及市场前景[J]. 广州化工,2008,36(2):18-22.
[3]胡志鹏. 草甘膦生产工艺路线比较[J]. 化学工业,2008,26(2):31-35.
[4]王冲. 催化氧化法合成草甘膦研究[D]. 杭州:浙江大学,2006.
[5]Pinel C,Landrivon E,Lini H,et al. Effect of the nature of carbon catalysis on glyphosate synthesis[J]. Journal of Catalysis,1999,182:515-519.
[6]Hershman A. Process for producing N-phosphonomethyl glycine:US,3969398[P]. 1976-07-13.
[7]韦少平,张丽娟,李致宝,等. 活性炭催化氧气氧化双甘膦制备草甘膦[J]. 农药,2009,48(8):558-560.
[8]周曙光. 双甘膦催化氧化制草甘膦过程[D]. 上海:华东理工大学,2010.
[9]徐晓辉. 草甘膦及其铵盐制备新工艺[D]. 湘潭:湘潭大学,2010.
[10]Besson M,Gallezot P,Perrard A,et al. Active carbons as catalysts for liquid phase reactions[J]. Catalysis Today,2005,102-103:160-165.
[11]Pigamo A,Besson M,Blanc B,et al. Effect of oxygen functional groups on synthetic carbons on liquid phase oxidation of cyclohexanone[J]. Carbon,2002,40:1267-1278.
[12]Chou S K. Process for removing surface oxides from activated carbon catalyst:US,4624937[P]. 1986-11-25.
[13]Chou S K. Amine oxidation using carbon catalyst having oxides removed from surface:US,4696772[P]. 1987-02-29.
[14]徐晓辉. 草甘膦及其铵盐制备新工艺[D]. 湘潭:湘潭大学,2010.
[15]Franze J E. Process for producing N-phosphonomethyl glycine:US,3950402[P]. 1976-04-13.
[16]陈彰明,黄当睦,陈福星,等. 双甘膦催化氧化合成膦甘酸的研究[J]. 工业催化,1993(1):26-31.
[17]刘金红,黄艳芳,刘志军,等. 贵金属催化剂制备条件对氧化性能的影响[J]. 农药,2010,49(2):100-113.
[18]黄伟,贾艳秋,孙盛凯. Pd-C催化剂研究进展[J]. 工业催化,2006,14(10):6-11.
[19]Felthouse T R. Oxidation with encapsulated co-catalyst:US,4582650[P]. 1986-04-15.
[20]王冲,陈志荣,尹红. 催化氧化法合成草甘膦研究进展[J]. 农药,2005,44(9):385-387.
[21]Hitzler M,Thalhammer F,Hammer B. Method for producing N-phosphonomethyl glycine:US,6730813[P]. 2004-05-04.
[22]Martin Ramon J L,Madronero J M. Preparation of N-phosphonomethyl :US,glycine by oxidarion of N-phosphonomethyliminodiacetic acid:US,5179228[P]. 1993-01-12.
[23]Hershman A,Gross D E,Friedman R M. Oxidation with coated catalyst:US,4579689[P]. 1986-04-01.
[24]Leiber M A,Ebner J R,Wan K T,et al. Use of a supplemental promoter in conjunction with a carbon-supported,noblemetalcontaining catalyst in liquid phase oxidation reactions:US,6586621B2[P]. 2003-07-01.
[25]Ebner J R,Leiber M A,Wan K T,et al. Deeply reduced oxidation catalyst and its use for catalyzing liquid phase oxidation reactions:US,6603039B1[P]. 2003-08-05.
[26]Hodgkinson I. Manufacture of N-phosphonomethyl glycine and its salts:US,5500485[P]. 1996-03-19.
[27]Ebner J R,Leiber M A,Wan K T,et al. Deeply reduced oxidation catalyst and its use in preparing N-phosphonomethyl glycine compounds:US,7067693B1[P]. 2006-06-27.
[28]Riley D P,Rivers W J.Process for producing N-phosphonomethyl glycine:US,4853159[P]. 1989-08-01.
[29]Riley D P,Fields D L,Rivers W. Homogeneous catalysts for selective molecular oxygen driven oxidative decarboxylations[J]. J. Am. Chem.Soc,1991,113(9):3371-3378.
[30]Riley D P. Process for producing N-phosphonomethyl glycine:EP,0314662B1[P]. 1992-11-19.
[31]Riley D P,Fields D L,Rivers W. Vanadium(Ⅳ,Ⅴ) salts as homogeneous catalysts for the oxygen oxidation of N-(Phosphonomethyl)iminodiacetic acid to N-(Phosphonomethyl)glycine[J]. Inorg. Chem. ,1991,30(22):4191-4197.
[32]石峰,陈天云,胡正辉,等. 氧气氧化法制备草甘膦[J]. 合肥工业大学学报,2008,31(8):1257-1259.
[33]Fields D L,Grabiak R C,Riley D P. Process for producing N-phosphonomethyl glycine:US,4898972[P]. 1990-02-06.
[34]Riley D P,Fields D L. Electron-transfer agents in metal-catalyzed dioxygen oxidations:Effective catalysts for the interception and oxidationofcarbonradicals[J]. J. Am. Chem. Soc.,1992,114(5):1881-1884.
