反相高效液相色谱法测定东方草莓中儿茶素的含量
2014-09-20,,,,,,,*
,,,,,,,*
(1.云南民族大学民族药资源化学国家民委-教育部重点实验室,昆明 650500;2.青海省食品药品检验所,西宁 810016)
反相高效液相色谱法测定东方草莓中儿茶素的含量
李蓉1,刘海青2,杨春涛1,江志勇1,郭俊明1,骆桂法2,黄相中1,*
(1.云南民族大学民族药资源化学国家民委-教育部重点实验室,昆明 650500;2.青海省食品药品检验所,西宁 810016)
建立东方草莓中(+)-儿茶素的含量测定方法。采用Agilent ZORBAX SB-C18色谱柱(4.6mm×150mm,5μm)分离;以乙腈-0.1%磷酸水为流动相进行梯度洗脱,流速为1mL/min,检测波长为280nm,对10批东方草莓中的(+)-儿茶素进行了含量测定。结果表明:(+)-儿茶素的保留时间为14.543min,其线性范围为0.0512~0.6400μg(R2=0.9999);加样回收率为99.70%(RSD 2.67%)。所建方法操作简单,稳定可靠,可用于东方草莓的质量评价。
东方草莓,(+)-儿茶素,反相高效液相色潽法,含量测定,质量控制
东方草莓FragariaorientalisLozinsk.为蔷薇科(Rosaceae)草莓属植物,俗称野草莓,为药食同源性药材。在我国主要分布于青海、甘肃、四川、陕西、山西以及东北、华中各省,朝鲜、蒙古及苏联远东地区也有分布[1]。东方草莓藏药名为“志达萨增”,干燥全草入药,常用于血热性化浓症,肺胃瘀血,黄水病脓疡[2],为藏药经典验方“七十味珍珠丸(然钠桑培)”[3]、“二十五味珍珠丸”[4]、“骨质增生乙组合(压迫神经引起疼痛)”、“八味秦皮丸”[5]等的重要成分。
长久以来,东方草莓作为藏药中的常用重要品种,缺少统一的质量标准衡定其质量,为保证其用药安全与药效,本课题组对东方草莓的质量进行了深入研究。目前尚未见有关东方草莓化学成分的具体报道。本课题组对其进行化学成分研究,经分离、鉴定发现(+)-儿茶素为东方草莓中的一主要化学成分。(+)-儿茶素具有抗病毒、抗癌、降血糖、抗菌、清除自由基等作用[6-10]。因此,可将(+)-儿茶素作为东方草莓中的特征性成分。
近年来,已有文献报道用高效液相法测定绿茶、杜仲、苦楝皮中儿茶素的含量[11-15],但目前未见有关东方草莓中儿茶素含量测定的相关报道。本实验用高效液相色谱法建立了东方草莓中(+)-儿茶素的含量测定方法,为东方草莓的质量控制研究提供科学依据。
1 材料与方法
1.1材料与仪器
(+)-儿茶素对照品 中国药品生物制品鉴定所提供,批号为11877-201203,纯度为97.20%;药材 本实验中使用了7个产地的10批药材,见表1;乙腈、甲醇 色谱纯;磷酸 分析纯;水 超纯水。
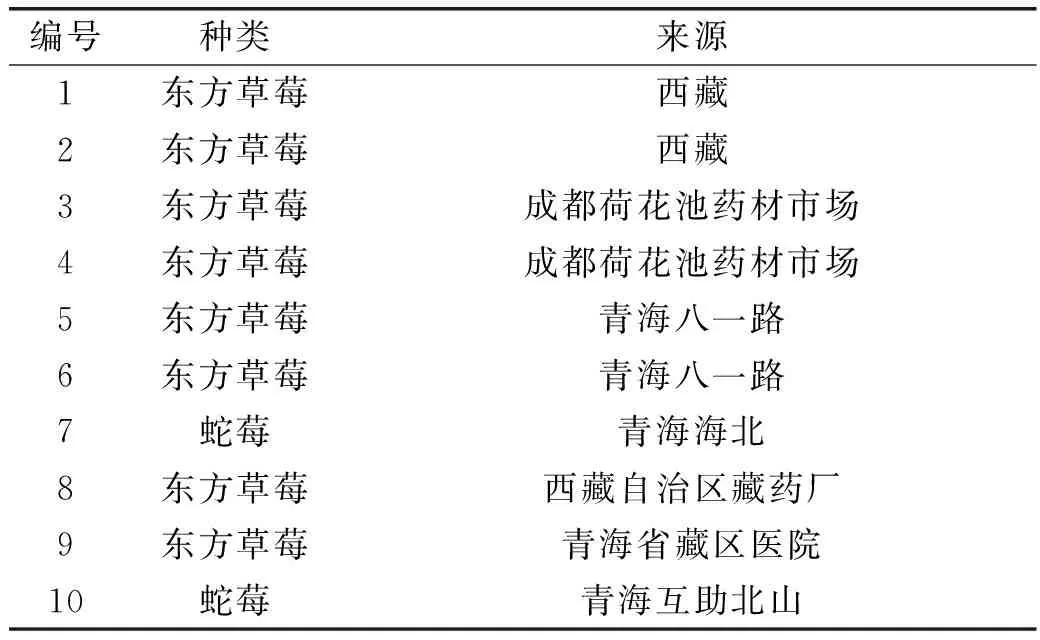
表1 10批不同来源的东方草莓药材Table 1 F. orientalis Lozinsk from 10 different sources
Agilent 1200高效液相色谱仪、Agilent ZORBAX SB-C18色谱柱(4.6mm×150mm,5μm) 安捷伦科技有限公司;DIONEX ULtiMate 3000高效液相色谱仪 戴安公司;Waters XTerra® RP18色谱柱(5μm,3.9×150mm) Waters公司;S105DU电子分析天平、AR224CN电子天平 瑞士梅特勒公司;AS20500A数控超声波清洗器 云南科仪玻有限公司;JP-150A-8高速多功能粉粹机 永康市久品工贸有限公司;DW100.P实验室超纯水机 上海和泰仪器有限公司。
1.2实验方法
1.2.1 对照品溶液的制备 精密称取(+)-儿茶素对照品1.28mg,置于50mL容量瓶中,加甲醇-水(1∶1)超声溶解,定容,摇匀,过0.45μm的滤膜,即得 0.0256mg/mL的对照品溶液。
1.2.2 供试品溶液的制备 经优选后的供试品溶液制备方法:取本品粉末0.30g,精密称定,置具塞锥形瓶中,精密加入甲醇-水(1∶1)10mL,密塞后称定此时具塞锥形瓶重量,放置1h,超声处理20min,放冷,再称定质量,用甲醇-水(1∶1)溶液补足减失的重量,摇匀,过滤,取续滤液过0.45μm的滤膜,即得供试品溶液。
1.2.3 色谱条件 Agilent ZORBAX SB-C18色谱柱(4.6mm×150mm,5μm),柱温:35℃;流动相乙腈(A)-0.1%磷酸(B),梯度洗脱(0~6min,6% A;6~20min,6%~8% A;20~ 25min,8%~10% A);检测波长280nm;流速1.0mL/min。
1.2.4 加样回收率测定样品的制备 精密称得(+)-儿茶素对照品2.45mg,置10mL容量瓶中,加甲醇-水(1∶1)溶液适量,超声溶解并稀释至刻线,摇匀即得对照品溶液(浓度为0.2450mg/mL)。取已知含量的东方草莓样品(8号)6份,每份0.30g,精密称定,置具塞锥形瓶中,精密加入甲醇-水(1∶1)溶液9mL、浓度为0.2450mg/mL的对照品溶液1mL,共10mL溶液,密塞后称定此时具塞锥形瓶重量,按照1.2.2方法制备供试品溶液。
2 结果与分析
2.1提取工艺的确定
对提取溶剂的考察时,考虑到东方草莓中鞣质丰富,在文献[11-13,15-19]的基础上,本实验不仅用水、乙醇-水(1∶1)、甲醇-水(1∶1)、乙醇和甲醇作为提取溶剂,还用了丙酮和乙醚,结果发现丙酮和乙醚在回流和超声条件下几乎不能将东方草莓中的(+)-儿茶素溶出,乙醇-水(1∶1)和甲醇-水(1∶1)对(+)-儿茶素的提取效率较好且相当,从节省经济考虑,选择甲醇-水(1∶1)为最佳提取溶剂。考察了超声和回流2种提取方法,结果表明两者的差别不大。由于儿茶素有4个同分异构体,长时间受热易发生差向立体异构化反应,转变为其同分异构体[20],因此,本实验用室温超声提取,以避免加热对样品测定的影响。对药材的浸泡时间和超声时间进行了考察,结果表明,药材用甲醇-水(1∶1)浸泡60min后室温超声提取20min,(+)-儿茶素的提取率较高。实验结果详见表2。

表2 东方草莓中(+)-儿茶素的提取工艺Table 2 Extraction process of(+)-catechin in F. orientalis Lozinsk
2.2色谱条件的选择
2.2.1 波长的选择 儿茶素类化合物属典型的黄烷醇类化合物[10],具有两个吸收带,即E2带和B带,分别在210nm左右和270~280nm处[21]。本实验曾采用双波长(210nm和280nm)检测,但由于210nm作为检测波长时基线漂移严重,所以选用280nm单波长检测。
2.2.2 流动相的选择 参考文献[11-14,19],本实验曾采用甲醇-0.1%磷酸水(10∶90)、乙腈-0.1%磷酸水(8∶92)等度洗脱,从供试品的分离情况和出峰时间等综合分析,最后选择以乙腈-0.1%磷酸水按1.2.3项下优选的色谱条件可保证分离度并避免干扰。
2.3标准曲线的建立
精密吸取以上对照品溶液2、10、15、20、25μL进样,按照1.2.3项下的色谱条件进行分析。以对照品的进样量(μg)为横坐标(X),对照品峰面积为纵坐标(Y),进行回归处理,得回归方程:Y=510.23X-1.0499(R2=0.9999),线性范围为0.0512~0.6400μg。(见图1)

图1 (+)-儿茶素的标准曲线Fig. 1 Standard curve of(+)-catechin
2.4方法学考察
2.4.1 耐用性考察 选择不同型号的高效液相色谱仪(DIONEX ULtiMate 3000)、不同型号的色谱柱依照1.2.3项下优选的色谱条件进行耐用性实验。理论塔板数以(+)-儿茶素峰计算不低于8000,此时指标成分能与其他相邻峰分离,分离度>1.5。此时,对照品和供试品液相图谱见图2。

表3 精密度实验Table 3 Precision of the measured results

表4 重现性实验Table 4 Reproducibility of the measured results

表5 回收率实验Table 5 Recoveries of the measured results

图2 对照品溶液(A)及供试品溶液(B)的HPLC图Fig. 2 The HPLC chromatogran of the standard solution(A)and the sample solution(B)
2.4.2 精密度考察 吸取2.1下的对照品溶液(浓度为0.0256 mg/mL),按照1.2.3项下优选的色谱条件测定,连续进样5次,每次10μL,分别记录峰面积。RSD为0.30%,表明系统的精密度良好。结果见表3。
2.4.3 重现性考察 取8号来源的东方草莓6份,按1.2.2项下优选的提取工艺制备供试品溶液,分别依照1.2.3项下优选的色谱条件测定,进样量为10μL,计算药材中儿茶素含量。平均值为0.0493mg/mL,RSD为2.89%。结果见表4。
2.4.4 回收率的测定 取1.2.4项下供试品溶液依1.2.3项下优选的色谱条件分别测定,进样量为5μL,平均回收率为99.70%,RSD为2.67%。结果见表5。
2.4.5 稳定性实验 分别吸取同一供试品溶液,依照1.2.3项下优选的色谱条件,在0、2、5、8、12、15、20、24、38、45、50、65h进样10μL,测定检定成分峰面积。结果表明,供试品溶液8h内RSD为0.27%,12h内RSD为1.25%,24h内RSD为1.37%,65h内RSD为1.38%,供试品溶液在65h内稳定。结果见表6。
2.5样品的含量测定
取7个产地的10批药材粉末,每个样品平行2份,按照1.2.2优选的提取工艺制备供试品溶液,依1.2.3项下优选的色谱条件分别测定,进样量为5~30μL,测定结果见表7。

表6 稳定性实验Table 6 Experiment results of the sample stability

表7 10批不同来源的东方草莓药材中(+)-儿茶素的含量Table 7 Content of(+)-catechin in F. orientalis Lozinsk. from 10 different sources
3 结论
本实验采用高效液相色谱法测定了东方草莓中(+)-儿茶素含量,分别考察了不同提取溶剂、提取方法及提取时间对东方草莓中(+)-儿茶素提取率的影响,得出东方草莓中(+)-儿茶素提取工艺(详见1.2.2项方法),并建立了色谱分析条件(详见1.2.3项方法)。该方法操作简单易行,精密度高,重现性和稳定性好,回收率为99.70%,可用于东方草莓中(+)-儿茶素含量测定。
经查阅文献[18],儿茶素类化合物在加热条件下不稳定。为保证测定结果的准确性,本实验所使用的对照品溶液现配现用(如果不能在当天完成分析测试,需将对照品溶液置于低温冰箱中保存,时间为2~3d)。通过对供试品溶液的稳定性考察,结果表明供试品溶液可在低温下存放2~2.5d;由于供试品的制备方法简便,测定周期短,建议现配制现用。
从表6的测定结果说明,蛇莓中的(+)-儿茶素含量明显高于东方草莓;不同产地的药材中(+)-儿茶素含量有差别,西藏地区>青海地区>成都地区。可见,(+)-儿茶素的含量与东方草莓生长的地理特征有着直接关系,并且海拔越高东方草莓中的(+)-儿茶素含量越高。
[1]吴征镒主编.中国植物志(37卷)[M].北京:科学出版社,2004:235.
[2]贾敏如,李星炜.中国民族药志要[M].北京:中国医药科技出版社,2005:281.
[3]加永泽培,杜元灏.藏药七十味珍珠丸对大鼠脑缺血梗塞面积的影响[J].西藏医药杂志.2012,33(3):56-57.
[4]王智森,赵献超,王凯梅,等.二十五味珍珠丸治疗脑出血后遗症临床疗效观察[J].中西医结合研究,2013,8,5(4):172-175.
[5]中华人民共和国卫生部药品标准[S].藏药第一册.1995:241.
[6]孙文基,绳金房.天然活性成分简明手册[M].北京:中国医药科技出版社,1998:109.
[7]曾亮,黄建安,李赤翎,等.儿茶素的抑菌效果及机理研究[J].食品工业科技,2009,30(5):89-92.
[8]Jankun J,Selman S H,Swiercz R,etal. Why drinking green tea could prevent cancer[J]. Nature,1997,387(5):561-561.
[9]Sakanaka S,Aizawa M,Kim M,etal. Inhibitory effects of green tea polyphenols on growth and cellular adherence of an oral bacterium,Porphyromonas gingvalis[J]. Biosci Biotechnol Biochem.,1996,60(5):745-749.
[10]刘超,陈若芸.儿茶素及其类似物的化学和生物活性研究进展[J].中国中药杂志,2004,29(10):1017-1021.
[11]Tetsuhisa G,Yuko Y,Masaaki K,etal. Simultaneous analysis of individual catechins and caffeine in green tea[J]. Journal of Chromatography A,1996,749:295-299.
[12]Henning S M,Claudia F L,Lee H W,etal. Catechin content of 18 teas and a green tea extract supplement correlates with the antioxidant capacity[J]. Nutrition and Cancer,2003,45(2):226-235.
[13]Chen Q S,Zhao J W,Chaitep S,etal. Simultaneous analysis of main catechins contents in green tea(Camellia sinensis(L.))by Fourier transform near infrared reflectance(FT-NIR)spectroscopy[J]. Food Chemistry,2009,113:1272-1277.
[14]曹丹,杨莉,侴桂新,等.苦楝皮中儿茶素的含量测定研究[J].中国药学杂志,2010,45(17):1035-1037.
[15]贾智若,李兵,李耀华,等.反相高效液相色谱法测定杜仲中儿茶素的含量[J].中国实验方剂学杂志,2013,19(5):117-119.
[16]夏晶,夏斌峰,季申. 反相高效液相色谱法测定拳参中儿茶素的含量[J].中国中药杂志,2007,32(11):1103-1104.
[17]王丽娟,张凤有.HPLC法测定蒙成药亚顺-12中的儿茶素的含量[J].中国民族医药杂志,2012,6:53-54.
[18]王钢力,于健东,田金改,等.儿茶药材化学成分分析——反相高效液相色谱法测定儿茶中的儿茶素和表儿茶素的含量[J].药物分析杂志,1999,19(2):88-90.
[19]潘金火,严国俊,卢欢.反相HPLC测定金荞麦药材和饮片中表儿茶素的含量.中国药学杂志,2010,45(14):1093-1096.
[20]陆蕴如主编.中药化学[M].北京:学苑出版社,1997:135-136.
[21]戴军,王洪新,陈尚卫,等.茶叶及茶多酚中儿茶素的高效液相色谱分析方法研究[J].色谱,2001,19(5):398-402.
Content determination of catechin inFragariaorientalisLozinsk. by RP-HPLC
LiRong1,LiuHai-qing2,YangChun-tao1,JiangZhi-yong1,GuoJun-ming1,LuoGui-fa2,HuangXiang-zhong1,*
(1. Key Laboratory of National Medicine Supported Jointly By State Ethnic Affairs Commission and Ministry of Education,Yunnan University of Nationalities,Kunming 650500,China;2. Qinghai Provincial Institute For Food and Drug Control,Xining 810016,China)
The purpose was to establish the method of(+)-catechin inFragariaorientalisLozinsk. The samples were performed on Agilent ZORBAX SB-C18(4.6mm×150mm,5μm)column,eluted gradiently with mixture of acetonitrile-0.1% phosphate solution at a flow rate of 1mL/min,and detected at 280nm. With this method,(+)-catechin in 10 batches ofF.orientalisLozinsk. was determined. Results showed that(+)-catechin retention time was 14.543min. The calibration curves had good linearity within the range of 0.0512~0.6400μg(R2=0.9999). The average recovery of(+)-catechin was 99.70%(RSD 2.67%). The method was easy and stable,and could be used for the quantitative analysis ofF.orientalisLozinsk.
FragariaorientalisLozinsk.;(+)-catechin;assay;quality control
2013-12-25 *通讯联系人
李蓉(19 -),硕士研究生,主要从事天然药物活性成分及质量控制研究。
国家药典委员会项目(M11);国家自然科学基金项目(21262047);云南民族大学传统傣药药用物质基础研究创新团队。
TS207.3
A
1002-0306(2014)17-0000-00
10.13386/j.issn1002-0306.2014.17.001
