NTO结晶形貌的预测
2014-09-18袁俊明刘玉存常双君王建华于雁武
马 松,袁俊明,刘玉存,常双君,王建华,于雁武
(中北大学化工与环境学院,山西 太原 030051)
引 言
3-硝基-1,2,4-三唑-5酮(NTO)是一种性能优良的高能钝感炸药[1],其爆轰性能和感度大小与其晶体形貌有一定的关系。NTO分子具有偶极子的特性,其结构的共价键具有方向性和饱和性,生长基元在结晶方位和晶体各个面族上附着的稳定性影响到结晶形貌[2],且NTO晶貌特征还与生长时的物理、化学条件有关,因此研究NTO晶体形貌需要把晶体内部结构同生长条件密切结合起来。目前关于晶体生长理论和晶貌预测的研究很多,如Singh M R[3]等人根据邻苯二甲酸氢钾其晶体可能的生长面及其面间距,用群体平衡模型(M-PBM)推出 KHP的晶体形貌;WANG Yan-chao[4]等人基于粒子群优化算法,计算出单晶硅具有低自由能的生长面,并根据其对称性信息和各晶面自由能预测了硅的生长晶习;Ulrich J[5]等人通过修正附着能法预测了苯甲酸在水中的重要生长面、重结晶形貌,其结果得到了实验验证;李鑫[6]等人结合实验与模拟计算,研究了添加剂对重结晶HMX晶体形貌控制的影响。
为了研究NTO晶貌形成的内在机理、微观尺度上溶剂分子与NTO晶面的作用机制和对其结晶形貌的影响,首先用附着能(AE)模型、分子动力学(MD)方法对NTO在真空、水及甲醇中的吸附能和晶体形貌进行了研究,并从NTO分子的极性、与溶剂所成氢键的角度分析了控制晶体形貌的内在和外在因素,为NTO重结晶工艺中溶剂的选择提供参考。
1 真空中NTO晶貌的预测
1.1 AE晶体生长模型
晶体生长的附着能(AE)模型是在周期性键链(PBC)模型的基础上发展起来的,PBC模型假设晶体表面成键所需时间与键合能成反比,晶面生长速度与键合能成正比,晶体结构由PBC组成,且晶体生长最快的方向是化学键最强的方向。而AE模型在此基础上定义了晶片能Ea为生长出一层厚度为dhkl的晶片释放出的能量,附着能Eatt为晶片附着在晶体表面(hkl)所释放出的能量,两者之和为晶体的晶格能Elatt[6-9]。

对于有机分子,晶格能可通过非成键原子间作用力函数和分子间作用综合计算求得,对于晶片能和附着能,可由中心分子与晶片内、外的所有分子总作用力计算得出。
Berkovitch-Yellin[10]对上述方法进行了改进,指出各晶面的相对生长速率(Rij)正比于附着能Eatt。

最低附着能的晶面生长得最慢,所以具有最大的形态学特征,从而决定了晶体的微观形貌。该方法是基于晶体对称性、分子间键链特性和晶面附着能来计算、推出有机分子在真空和有机溶剂中的附着能、晶貌特征。
1.2 NTO晶体模型的构建
在CCDC剑桥晶体结构数据库中可查出计算所需的NTO晶胞参数,共有α和β两种晶型,因后者密度较高,在起爆药内应用较多,故以后者为研究对象,β型NTO晶体为单斜晶系,属于CS点群,其晶胞参数为:a=9.322,b=5.4791,c=9.0685,α=γ=90°,β=101.322°。
根据β型NTO晶体中原子分数坐标和对称性信息在material studio 6.0中建立原胞,并在Discover模块下进行5000步的几何优化,用优化后的原胞建立3×3×3的超晶胞,为进一步消除NTO超胞中不合理的能量和构象,用Forceit模块下退火(Annealing)操作对其进行300~500K升、降温循环处理,此过程总步数为10 000步,每隔1 000步保存一次构象,采用Compass力场,NPT系综,Andersen控温方法,各原子起始速度按Maxwell分布取样,Velocity Verlet算法标定原子速度,分别用Atom based和Ewald方法计算范德华和静电作用,原子截断半径取9.5Å,缓冲宽度取0.05[11]。经此过程可得到结构合理、势能较低的构象文件,选取最低势能构象文件作后续计算,图1为优化后的NTO超晶胞。
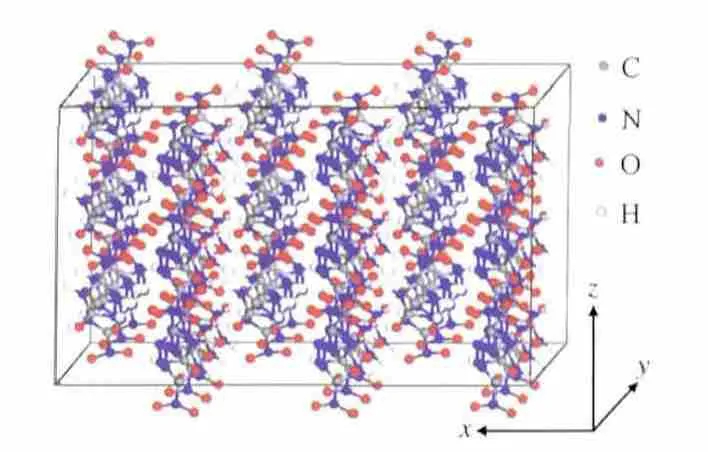
图1 优化后的NTO超晶胞Fig.1 The optimized supercell of NTO
1.3 真空中NTO的晶貌
预测NTO真空中晶貌的主要目的是从晶体的周期性键链(即分子间作用)和自身附着能的角度分析无溶剂影响下晶貌形成的内部因素,在此基础之上用晶面与溶剂分子的吸附能修正NTO晶体重要生长面的附着能,以此预测和研究NTO在水及甲醇中的晶貌。
在Morphology模块下,用1.2节中的力场类型、热力学系综、截断半径等控制方法,对所得豫弛后的构象用附着能法预测NTO晶体形貌,结果见图2(a),用Crystal Graph法计算优化后NTO原胞内分子基团间的相互作用,见图2(b),虚线为分子间力,强弱为蓝>紫>红(蓝线方向的晶面生长最快,已消失),NTO重要晶面的生长信息见表1。

图2 真空中NTO晶体形貌(a)和分子间作用力(b)Fig.2 Morphology(a)and molecular interaction(b)of NTO in vacuum

表1 真空中NTO重要晶面的生长信息Table 1 Growth of important surface of NTO in vacuum
从图2可见,NTO分子在(010)方向形成了一种偶极生长基元,-NO2基团电子云密度大,为负极面,与三唑环上的正电荷中心(距离C原子较近)有很强的分子间力,见表1中的Eatt(Elec),(010)晶面的法线与-NO2基团伸展方向夹角小,受此极化作用影响,分子在(010)面附着效率较高,故(010)面的生长速率较(100)和(001)面快,且晶面面积较小,相对于其他快速生长、或已消失的面仍顽强显露。
(001)面的法线几乎垂直于-NO2基团和三唑环所在平面,分子的附着主要靠共轭体系间微弱的范德华力作用,晶面生长速率较(010)面低且显露面积大;(100)面法线亦与-NO2基团伸展方向夹角大且平行于三唑环共轭平面,另外NTO分子在此方向上侧向排布,附着能主要由羰基与三唑基团间的亲和作用提供,致使分子的附着效率最低,其晶面所占面积最大,成为NTO形态学上最重要的生长面。
相对于已消失、或显露面积小的生长面,(010)、(100)和(001)晶面为重要生长面,因Rij∝Eatt,故真空中各晶面生长速率(010)≫(100)≈(001),其形貌为长四棱柱。据分析可知,真空中NTO晶体形貌主要受其分子环上键链作用弱的(100)和(001)晶面决定,其次是极性较强的(010)晶面。
2 溶剂中NTO晶貌的预测
2.1 吸附模型的构建与优化
溶剂分子对NTO晶貌的影响是通过与其晶面的吸附作用实现的,故需建立水、甲醇与NTO晶面的吸附模型,再求解其吸附能。水和甲醇分子的模型由Sketch建立后,也采用1.2节中方法进行几何构型和能量的豫弛。
由表1可知,(010)、(001)和(100)晶面为三组重要生长面,其面积占晶体总表面积的92.78%,为减小计算量,只计算这三组晶面对水和甲醇分子的吸附能。
对退火所得的3×3×3超晶胞(010)、(001)和(100)面进行切割,切割厚度为一个分子层,再对此二维结构加20Å的真空层,由此形成了具有吸附面的层晶。分别往各个吸附面加入水和甲醇分子,用文献[12]中的方法调整合理的构型,以形成6个初始吸附模型,对每种模型进行几何优化,方法与前述相同。经优化后的水、甲醇与NTO各晶面的吸附模型见图3(虚线为氢键),其中(100-H2O)吸附模型与文献[2]中的NTO与水全优化构型相符。
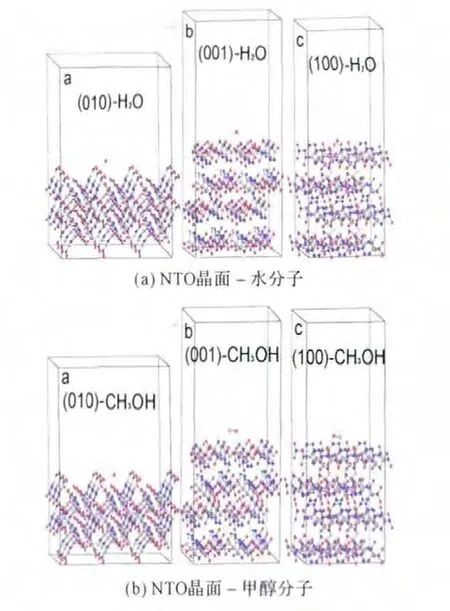
图3 NTO晶面与水分子(a)和甲醇分子(b)的吸附模型Fig.3 Sorption models of NTO crystal surfaces with water(a)and methanol(b)
2.2 吸附过程的动力学计算
固定除表层以外的分子层以减少计算量(溶剂分子与固定层距离大于9.5Å),用Forceit模块下的动力学过程对优化的6种吸附模型进行NPT系综下的MD计算,总时间为150ps,步长为1fs,其他控制参量与退火过程相同,MD过程结束后,用各模型能量稳定后的构型(后50帧)分别对整个体系、溶剂分子和晶面的能量求均值,再用吸附能计算公式(3)求解吸附能[6-9],计算结果见表2。

式中:Etotal为吸附体系的总能量(kJ/mol);Esolv为溶剂分子单点能(kJ/mol);Esurf为晶面的表面能(kJ/mol)。
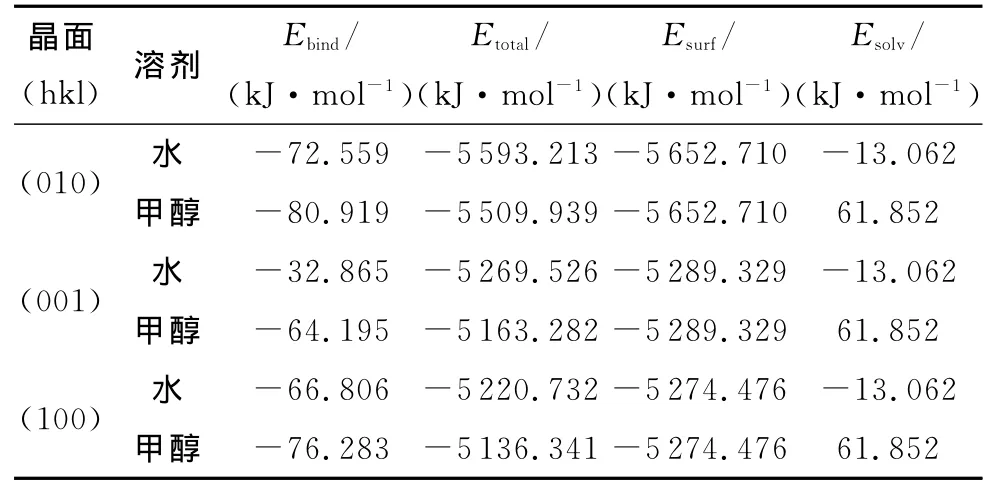
表2 各晶面与溶剂分子的吸附能Table 2 The sorption energy between surface and solvent
2.3 溶剂中NTO的晶貌
为研究溶剂及其他因素对NTO晶体形貌的影响,需要用表2中的吸附能对表1中的附着能进行修正(本研究仅考虑溶剂因素)。因各溶剂对晶体不同NTO晶面的附着能影响不同,可用重要生长面(010)、(001)和(100)对溶剂分子(水和甲醇)的吸附能对此三组晶面上NTO分子的附着能予以修正,计算公式为[8-9]:

式中:Eatt′为修正后的附着能(kJ/mol);Ebind为晶面对溶剂分子的吸附能(kJ/mol);Eatt为真空中的附着能(kJ/mol)。另外由AE模型理论可推断出修正后的生长速率、面心距和附着能亦符合R′ij∝D′ij∝E′att,三者的计算结果见表3,可见溶剂分子对晶貌的影响是通过与晶面的吸附来影响NTO分子在各面的附着能量与效率,溶剂与面相吸附能愈大,面生长愈慢,反之亦然。
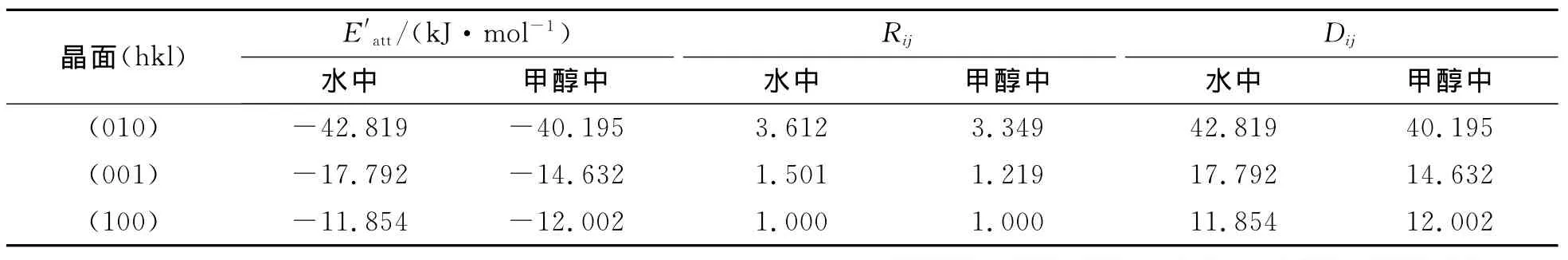
表3 水和甲醇中NTO主要晶面的附着能Table 3 Attachment energy of important surface of NTO in water and methanol
根据水、甲醇与不同晶面吸附能,修正各晶面的附着能值,为各晶面指定相对生长速率和中心到面的距离,再通过Wulff图对以上信息进行分析,推导出晶体形态,并与文献[13]中NTO在水和甲醇中的结晶形态电镜扫描图进行对比,结果见图4和图5。

图4 NTO 在水中的晶貌预测(a)和SEM 图(b)[13]Fig.4 Prediction of morphology on NTO in water(a)and SEM image(b)

图5 NTO在甲醇中的晶貌预测(a)和SEM 图(b)[13]Fig.5 Prediction of morphology on NTO in methanol(a)and SEM image(b)
据表1~表3可知,NTO各晶面会通过对水和甲醇分子的吸附而降低自身的附着能。文献[6,8-9]亦显示,极性溶剂与有机分子的相互作用中,氢键的成键强度较范德华力和静电力显著。
综合图4和表3可知,(010)晶面上的硝基分布密集,水分子中的氢易于硝基氧缔合成氢键,且成键密度较高,所以该面对水分子吸附能最大,即水分子对(010)面附着能的抑制最明显;水分子中的氢、氧原子能与NTO分子上的9位氧、11位氢原子[2]间形成两个氢键且方向垂直于(100)面,故与(100)面吸附能也较强,但该面在真空中的附着能基数较小,水的抑制作用较(010)面次之;(001)面与水的吸附能由氢键和范德华力共同作用(该面上氢、氧原子分布少,氢键对吸附能贡献小),且量值最小,水分子对附着能抑制作用最小。故水中各面生长速率为(010)>(001)>(100),其结晶形态为扁平的六面体。
从图5和表3可见,甲醇分子同各面的吸附机理类似于水,但由于甲基推电子集团使羟基周围电子云密度高,分子偶极距大[7-9],与各面形成的氢键作用较强且键能差距较小,致使(010)和(001)面的附着能降低的量值相近;另外在(100)面所成的氢键无水分子与该面所成氢键的特殊性,故对(100)面的抑制强度与水相当。故甲醇中各面的生长速率(010)>(100)≈(001),其结晶形态为四棱柱。
总之,溶剂分子对晶貌的影响由NTO分子内氧原子的排布、数目,溶剂分子偶极距、羟基数目所决定。用修正后的AE模型所预测的晶貌与文献[13](P-84)中扫描电镜所观测的实际晶貌能较好地匹配,说明预测是较成功的。
3 结 论
(1)利用分子动力学和修正附着能的方法可对NTO晶体在真空、水和甲醇环境中的晶貌进行预测,其结果和文献中的扫描电镜图能较好地匹配。
(2)真空中,NTO晶体的主要生长面为(010)、(100)和(001)面,因附着能随晶面法向极性的增强而升高,故非极性面(100)和(001)生长最慢,有硝基显露的极性面(010)次之。
(3)水和甲醇通过与NTO分子环上的硝基或羰基氧形成的氢键而降低各晶面附着能,且硝基与醇羟基所成氢键最强。表现为水中各面生长速率(010)>(001)>(100),甲醇中(010)>(100)≈(001),且总体上甲醇中的生长速率稍小。
[1] Nandi A K,Singh S K,Kunjir G M,et al.Assay of insensitive high explosive 3-nitro-1,2,4-triazol-one(NTO)by acid-base titration [J].Central European Journal of Energetic Materials,2013,10 (1):113-122.
[2] 方国勇,徐丽娜,肖鹤鸣,等.3-硝基-1,2,4-三唑-5-酮与NH3及H2O分子间相互作用的理论研究 [J].化学学报,2005,63:1056.FANG Guo-yong,XU Li-na,XIAO He-ming,et al.Theoretical study on intermolecular interactions of 3-nitro-1,2,4-triazol-5-one with NH3and H2O [J].Acta Chimica Sinica,2005,63:1056.
[3] Singh M R,Ramkrishna D.A comprehensive approach to predicting crystal morphology distributions with population balances [J].Cryst,Growth Des,2013,13:1397-1411.
[4] WANG Yan-chao,LüJian,ZHU Li,et al.Crystal structure prediction via particle-swarm optimization[J].Physical Review B,2010,82:094116.
[5] Ulrich J.Predicting crystal morphology grown from solution [J].Chem, Eng, Technol,2012,35 (6):1009-1012.
[6] 李鑫,陈树森,李丽洁,等.添加剂对HMX重结晶晶体形貌的影响 [J].火炸药学报,2011,34(3):15-19.LI Xin,CHEN Shu-sen,LI Li-jie,et al.Influences of additives on HMX crystal morphology [J].Chinese Journal of Explosives and Propellants,2011,34(3):15-19.
[7] ZHOU Kun,LI Jun,LUO Jian-hong,et al.Crystal growth,structure and morphology of rifapentine methanol solvent [J].Chinese Journal of Chemical Engineering,2012,20(3):602-607.
[8] CHEN Jian-xin,WANG Jing-kang,ZHANG Ying,et al.Crystal growth,structure and morphology of hydrocortisone methanol solvate[J].Journal of Crystal Growth,2004:265-273.
[9] 陈建新,王静康,尹秋响,等.氢化可的松形貌预测[J].天津大学学报,2006,39(增刊):3-6.CHEN Jian-xin,WANG Jing-kang,YIN Qiu-xiang,et al.crystal morphology prediction of hydrocortisone.[J].Journal of Tianjin University,2006,39(Suppl):3-6.
[10]Berkovitch-Yellin Z.Toward an ab initio derivation of crystal morphology[J].J Am Chem,Soc,1985:107.
[11]赵树森,付一政,梁晓艳,等.HTPB/增塑剂共混物的介观动力学模拟 [J].高分子材料科学与工程.2011,27(5):186-187.ZHAO Shu-sen,FU Yi-zheng,LIANG Xiao-yan.et al.Mesoscale dynamic simulation on HTPB/plasticizers blends[J].Polymer Materials Science and Engineering,2011,27(5):186-187.
[12]王志明.羟基磷灰石晶习及降解特性的研究 [D].天津:天津大学,2007.
[13]Teipel U.Energetic Materials[M].Weinheim:Wiley-Vch Verlag GmbH &Co.KGaA,2005:84-139.
