NEPE推进剂/衬层结构-性能MD模拟(Ⅱ)
——复杂体系组分分子迁移和配方设计示例①
2014-09-07刘冬梅肖继军池旭辉庞爱民肖鹤鸣
朱 伟,刘冬梅,肖继军,池旭辉,庞爱民,肖鹤鸣
(1.南京理工大学 化工学院,分子与材料计算研究所,南京 210094; 2.嘉兴学院 生物与化学工程学院,嘉兴 314001;3.中国航天科技集团公司四院四十二所,襄阳 441003)
NEPE推进剂/衬层结构-性能MD模拟(Ⅱ)
——复杂体系组分分子迁移和配方设计示例①
朱 伟1,2,刘冬梅1,肖继军1,池旭辉3,庞爱民3,肖鹤鸣1
(1.南京理工大学 化工学院,分子与材料计算研究所,南京 210094; 2.嘉兴学院 生物与化学工程学院,嘉兴 314001;3.中国航天科技集团公司四院四十二所,襄阳 441003)
用分子动力学(MD)方法,对(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI复杂的推进剂/衬层模型体系进行295 K-NVT模拟研究,展示了组分分子的浓度分布和迁移状况,发现HMX和NPBA分子有向界面层迁移趋势,而AP则呈平均分布态势。以RDX等量取代HMX后所得新配方的MD模拟研究表明,前者拉伸模量(E)、体模量(K)和剪切模量(G)、柯西压(C12-C44)和K/G值均有明显下降,表明新配方的刚性、强度和延展性均有下降;新配方中引发键(N—NO2)最大键长(1.528 Å)明显大于原配方中相应值(1.503 Å),预示新配方感度增大、安全性将下降;比较RDX、HMX与其他组分之间的结合能,前者小于后者,预示新配方的相容性较差。
推进剂/衬层界面;组分迁移;力学性能;感度;相容性;分子动力学模拟
0 引言
相容性、安全性和和力学性能是推进剂、衬层及其界面所需重要性能,直接影响高能固体发动机的工作效能和寿命长短。这些性能一般依靠实验测定,如能通过理论模拟和计算加以定量预测或定性评估,则不仅可节省人力、物力和财力,缩短研究周期,而且可指导配方设计,是重大基础研究所迫切需要解决的重要课题。
近年来兴起的分子动力学(MD)模拟,通过对庞大复杂体系结构和性能的模拟计算,正好满足为高能复合材料的配方设计提供丰富的信息、规律和理论指导的需求。通过MD模拟研究固体推进剂和衬层的工作已有一些,研究大都集中在推进剂中粘接剂与增塑剂的相容性和力学性能[1-8],推进剂中可迁移组分的影响作用[9-10]及推进剂颗粒填充模型建立[11]。但这些工作主要集中于双组分体系,个别多组分体系也只限制在推进剂中的粘结与增塑体系中的简单模型[7]。因实际应用的固体推进剂均为多组分复杂体系,而对于推进剂/衬层复杂体系的MD模拟至今未见公开报道。
本文选择常用粘接剂聚乙二醇(PEG)、增塑剂硝化甘油(NG)和1,2,4-丁三醇三硝酸酯(BTTN)、聚合物键合剂(NPBA)、氧化剂奥克托今(HMX)和高氯酸铵(AP)以及推进剂和衬层固化体系PEG(聚乙二醇)/N-100(异氰酸酯)//HTPB(端羟基聚丁二烯)/TDI(甲苯二异氰酸酯)混合体系,即以推进剂/衬层(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI复杂体系为研究对象,搭建合理模型进行MD模拟,首次展示典型组分分子向界面迁移的有趣图像,并以RDX等量取代HMX,比较2种配方的相容性、力学性能和安全性(感度),提供MD模拟用于配方设计的范例。
1 模型构建和模拟细节
对于(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI推进剂/衬层十分复杂的混合体系,按一定配比和添加次序,由简到繁加以构建。
1.1 (PEG/NG/BTTN)/NPBA/HMX/AP混合体系模型搭建与MD模拟
在(PEG/NG/BTTN)/NPBA/HMX/AP体系中,共含10 015个原子,依据其配比和先后次序要求加以混合。即按照实验要求,先将NG和BTTN以1∶1的比例混合搭好模型,然后与PEG混合组成粘合剂;再将构建好的粘合剂体系与NPBA、HMX和AP分子,均匀地放入边长为200 Å×200 Å×200 Å的周期箱中。在修正和扩展的MPCFF力场[12]和NVT系综下进行20 ps MD模拟,以使体系达到平衡;然后,缓慢缩小周期箱体积,同时进行MD模拟使达到平衡;重复此过程,直到体系密度接近其理论值。取其最终结构,温度设定为295 K,再进行200 ps MD周期性模拟,直至获得平衡结构。图1(a)和(b)分别示出(PEG/NG/BTTN)/NPBA/HMX/AP混合体系的初始结构和平衡结构。
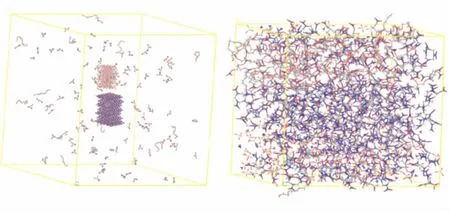
(a)初始结构 (b)平衡结构图1 (PEG/NG/BTTN)/NPBA/HMX/AP 混合体系初始结构和平衡结构
1.2 PEG/N-100和HTPB/TDI固化体系模型搭建与MD模拟
将2条端基饱和链节数为20的PEG高分子链与N-100构成的混合模型(如图2所示),先进行MM优化,然后在295 K-NVT系综下,进行200 ps的MD模拟,所得结构用作研究固化反应的初始模型。对此模型,先在323 K-NPT系综下进行100 ps的MD模拟,然后使用热力学方法、手动连接邻近的两分子—NCO与—OH活性基团,完成反应[13]。对最后所得混合体系,在295 K-NVT系综下,再进行200 ps MD模拟。体系中含430个原子,PEG和N-100的质量分数分别为64.1%和35.9%。
图2 N-100和PEG混合模型
仿照PEG/N-100固化体系搭建方式与模拟方法,HTPB/TDI固化体系也进行类似的搭建与模拟。该固化体系中含有5条链节数为20端基饱和的HTPB分子链和5个TDI分子,共含1 145个原子,HTPB和TDI的质量分数分别为86.8%和13.2%。图3给出了HTPB/TDI混合模型。

图3 HTPB和TDI的混合模型
1.3 (PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI模型搭建与模拟
将得到的(PEG/NG/BTTN)/NPBA/HMX/AP平衡结构与PEG/N-100固化平衡结构,构成一个整体模型,共含10 445个原子。对此模型做周期性MD模拟,直到达到热力学平衡。再将获得的此含固化反应的推进剂模型结构,与HTPB/TDI衬层固化平衡结构构成一个整体模型,共含11 690个原子,类似地完成MD模拟,最终获得如图4所示推进剂/衬层平衡结构。

图4 (PEG/NG/BTTN)/NPBA/HMX/AP/PEG/ N-100//HTPB/TDI体系的平衡结构
对于以RDX等量取代HMX,模型搭建与模拟方法同HMX一样,为节省篇幅,不再赘述。图5给出了(PEG/NG/BTTN)/NPBA/RDX/AP/PEG/N-100//HTPB/TDI混合体系的平衡结构。

图5 (PEG/NG/BTTN)/NPBA/RDX/AP/PEG/N-100// HTPB/TDI体系的平衡结构
2 结果与讨论
2.1 组分分子的浓度分布和迁移状况
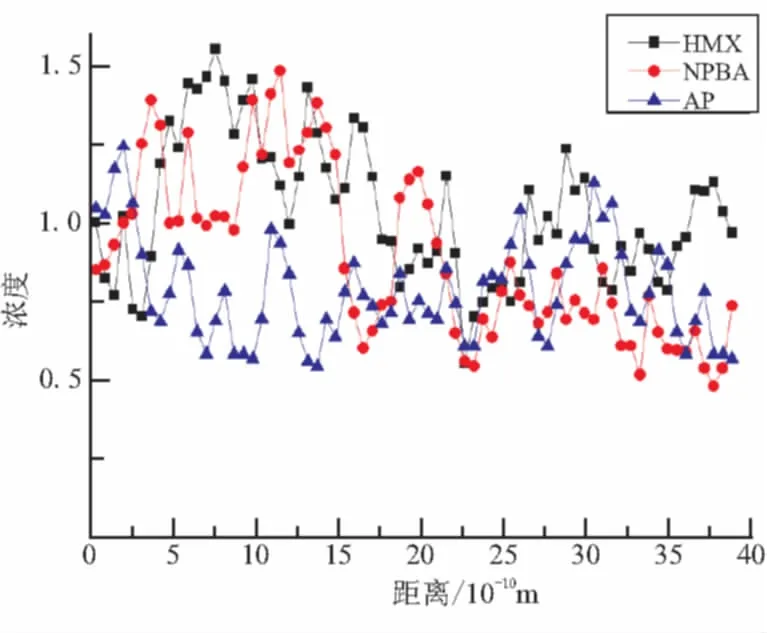
从图4多组分体系的平衡结构可见,在推进剂/衬层界面处,亦即在(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100与HTPB/TDI交界层,明显可见HMX分子的富集,并大致可推断HMX分子有向界面层迁移的趋势。为仔细探究组分分子的运动迁移状况,作为例子以下给出HMX、NPBA和AP分子在OA(X)、OB(Y)和OC(Z)方向上的浓度分布曲线,如图6所示。
因为推进剂/衬层之界面层在OC(Z)方向上,故可细致考察HMX、NPBA和AP分子在OC(Z)方向上的浓度分布图。从图6(c)可见,HMX和NPBA分子在6 Å以内分布较少,随着向界面层接近,HMX和NPBA分子的浓度迅速增加,在30 Å处,达到顶峰,而后又缓慢下降,直至减小为零。从HMX和NPBA分子的整体浓度分布图,可清晰看到HMX和NPBA分子有向界面层迁移的趋势。这与实验研究HMX有向界面迁移的结果一致[14]。而AP分子则基本上呈较平均分布的态势,并无明显迁移情况的方向性。
2.2 结合能和相容性比较
先前曾建议以组分之间的相互作用能(结合能)表征其相容性的优劣[6]。对于(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI复杂体系(记为配方I),可将(PEG/NG/BTTN)/NPBA/AP/PEG/N-100//HTPB/TDI视作一个整体,记作子体系1,其能量用E1表示;将HMX记为子体系2,其能量用E2表示。对于含RDX混合体系(记为配方II)也作类似处理。

(a)OA(X)方向
(a)OB(Y)方向

(c)OC(Z)方向图6 HMX、NPBA和AP分子在OA(X)、OB(Y)、OC(Z)方向的浓度分布
定义结合能Ebind是相互作用能Einter的负值,即Ebind=-Einter。则配方I中HMX与子体系之间的相互作用能为
Einter=ETotal-E(PEG/NG/BTTN)/NPBA/AP/PEG/N-100//HTPB/TDI-EHMX
这里ETotal(ET)、E(PEG/NG/BTTN)/NPBA/AP/PEG/N-100//HTPB/TDI(E1)和EHMX(E2)分别为混合体系、子体系(PEG/NG/BTTN)/NPBA/AP/PEG/N-100//HTPB/TDI和HMX的平均单点能。配方Ⅱ与此类似。计算结果见表1。

表1 2种不同配方体系的结合能
结合能愈大。表明形成的多组分混合体系越稳定,反映了各组分间的彼此相容性越好。所以,曾建议以结合能Ebind的大小来预示和比较高能体系的相容性[6-7,15]。从表1可见,体系Ⅰ的ET(306 573.3 kJ/mol)大于体系Ⅱ的ET(289 438.6 kJ/mol),即含HMX的体系Ⅰ整体的热力学稳定性较含RDX的混合体系Ⅱ稳定,且HMX与子体系1的Ebind(223 319.2 kJ/mol)明显大于RDX与其他子体系1的Ebind(202 257.5 kJ/mol),即含HMX配方的子体系之间的相容性优于含RDX配方的子体系之间的相容性。
2.3 力学性能比较
表2给出了基于MD模拟原子运动轨迹由静态力学分析方法[16],求得的含HMX配方体系Ⅰ和含RDX配方体系Ⅱ的力学性能。
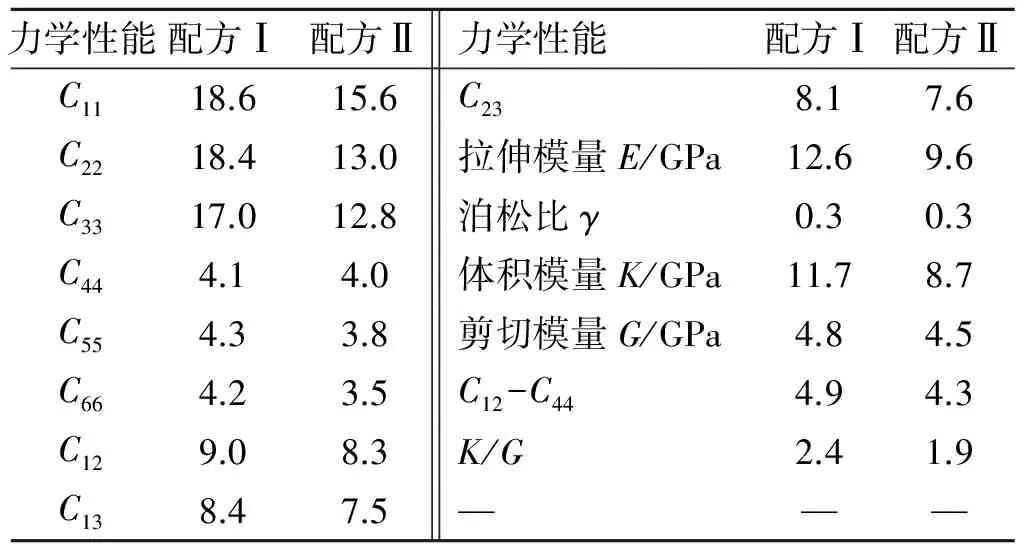
表2 2种不同配方的力学性能
从表2可见,氧化剂为HMX的配方Ⅰ的弹性系数、工程模量及柯西压(C12-C44)和K/G值等力学性能参数均高于含RDX的配方Ⅱ,表明前者的刚性和强度均较后者为强,且延展性也以前者为优。这就是说,若以RDX取代HMX,则推进剂/衬层体系(界面)的刚性、强度和延展性将减小,力学性能将变差。
2.4 安全性能比较
如何利用MD模拟结果关联感度特性(安全性),建议重点关注其中易爆燃组分,因为易爆燃组分的引发键是分子中最弱的化学键,在外界刺激下,将优先断裂、引发热解或起爆。所以,研究易爆燃组分的引发键很有必要。
表3给出了混合体系Ⅰ和混合体系Ⅱ中引发键的平均键长(Lave)和最大键长(Lmax)。为便于比较,表3中还列出纯NG、BTTN、HMX和RDX晶体的MD模拟所得Lave和Lmax。从表3可见,在混合体系中,由于存在多组分之间的相互作用,它们的Lave和Lmax与纯晶体中的相应值比较,均有所增大。例如,在含HMX混合体系中,其引发键(N—NO2)最大键长(Lmax)从纯晶体的1.486 Å增加到1.503 Å;而在含RDX的混合体系中,其引发键(N—NO2)的最大键长(Lmax)则由纯晶体的1.512 Å增加到1.528 Å。重要的是比较2种配方,NG和BTTN的O—NO2键长几无变化;但HMX引发键(N—N)的最大键长Lmax(1.503 Å)远小于RDX的相应值(1.528 Å)。这就是说,以RDX取代HMX后,新配方中引发键(N—NO2)的Lmax远大于原配方的相应值。以建议的引发键Lmax判据[17-20],可预测新配方的热和撞击等感度将大于原配方,这主要归因于RDX比HMX的感度大[21-22]。
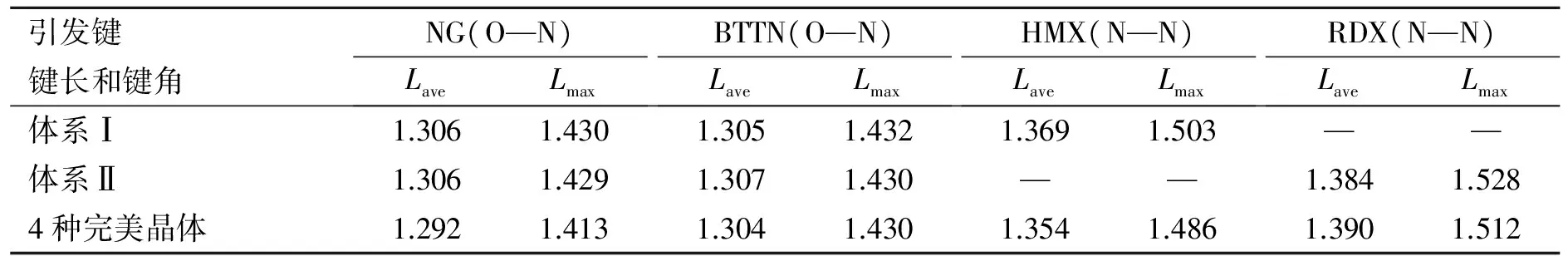
表3 2个配方中引发键的平均键长(Lave)和最大键长(Lmax)
3 结论
(1)搭建了(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI复杂的推进剂/衬层模型(配方I),并以RDX等量取代HMX,搭建相应的配方II模型。首次对2种复杂的配方体系进行了系统的MD比较研究。
(2)展示了配方I中组分分子的浓度分布和迁移状况,发现HMX和NPBA组分的分子有向界面层迁移的趋势,而AP分子则呈平均分布态势。
(3)配方II子体系之间的结合能比配方I小,表明相容性不如配方I;配方II工程模量减小,表明刚性和强度下降;柯西压(C12-C44)和K/G值亦减小,表明延展性亦减小;配方II中引发键N—NO2的Lmax增大,表明感度将增大,安全性下降。可见,含RDX的配方II没有含HMX的配方I好。本文首次提供了MD对复杂高能体系配方设计的指导作用。
[1] 姚维尚,李倩,谭惠民.NEPE推进剂粘合剂性能的分子模拟研究[J].含能材料,2007,15(6):650-655.
[2] 杨月诚,焦东明,强洪夫,等.HTPB推进剂组分溶度参数的分子模拟研究[J].含能材料,2008,16(2):191-195.
[3] 付一政,刘亚青,兰艳花.端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟[J].物理化学学报,2009,25(7):1267-1272.
[4] 赵贵哲,冯益柏,付一政,等.端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟和介观模拟[J].化学学报,2009,67(19):2233-2238.
[5] 焦东明,杨月诚,强洪夫,等.HTPB固体推进剂增塑剂选取分子模拟研究[J].化学研究与应用,2009,21(6):805-809.
[6] Xu X J,Xiao J J,Huang H,et al.Molecular dynamics simulations on the structures and properties ofε-CL-20-based PBXs[J].Sci.China Ser.B,2007,50(6):737-745.
[7] 于艳春,朱伟,肖继军,等.四组分高能体系结合能和力学性能的分子动力学模拟[J].化学学报,2010,68(12):1181-1187.
[8] 付一政,胡双启,兰艳花,等.HTPB/增塑剂玻璃化转变温度及力学性能的分子动力学模拟[J].化学学报,2010,68(8):809-813.
[9] 李红霞,强洪夫,武文明.丁羟推进剂黏结体系中增塑剂迁移的分子模拟[J].火炸药学报,2008,31(5):74-78.
[10] 李红霞,强洪夫,王广,等.基于MD方法的增塑剂扩散行为的模拟研究[J].含能材料,2009,17(1):36-41.
[11] 史佩,李高春,王玉峰,等.复合推进剂颗粒填充模型的分子动力学模拟方法[J].计算机与应用化学,2007,24(5):665-668.
[12] Zhu W,Wang X J,Xiao J J,et al.Molecular dynamics simulations of AP/HMX composite with a modified force field[J].Journal of Hazardous Materials,2009,167(1):810-816.
[13] 李红霞,强洪夫,武文明.HTPB与TDI固化的分子模拟研究[J].固体火箭技术,2008,31(6):602-606.
[14] 吴丰军,彭松,池旭辉,等.NEPE推进剂/衬层粘接界面XPS表征[J].固体火箭技术,2009,32(2):192-196.
[15] Xiao J J,Huang H,Li J S,et al.A molecular dynamics study of interface interactions and mechanical properties of HMX-based PBXs with PEG and HTPB[J].Journal of Molecular Structure:THEOCHEM,2008,851:242-248.
[16] Swenson R J.Comments on virial theorems for bounded systems[J].Am.J.Phys.,1983,51:940-942.
[17] 朱伟,肖继军,郑剑,等.高能混合物的感度理论判别——不同配比和不同温度AP/HMX的MD研究[J].化学学报,2008,66(23):2592-2596.
[18] Xiao J J,Zhao L,Zhu W,et al.Molecular dynamics study on the relationships of modeling,structural and energy properties with sensitivity for RDX-based PBXs[J].Sci.China Ser.B,2012,55:2587-2594.
[19] Xiao J J,Wang W R,Chen J,et al.Study on structure,sensitivity and mechanical properties of HMX and HMX-based PBXs with molecular dynamics simulation[J].Comput.Theor.Chem.,2012,999:21-27.
[20] Xiao J J,Li S Y,Chen J,et al.Molecular dynamics study on the correlation between structure and sensitivity for defective RDX crystals and their PBXs[J].J.Mol.Model,2013,19:803-809.
[21] 董海山,周芬芬.高能炸药及其相关性能[M].北京:科学出版社,1989:252-253,266-267.
[22] 肖继军,朱卫华,朱伟,等.高能材料分子动力学[M].北京:科学出版社,2013:120-129.
(编辑:刘红利)
Molecular dynamics simulation of the structures and properties of NEPE propellant/liner(Ⅱ) —Demonstrating component molecule migrating and formulation for complex system
ZHU Wei1,2,LIU Dong-mei1,XIAO Ji-jun1,CHI Xu-hui3,PANG Ai-min3,XIAO He-ming1
(1.Molecules and Materials Computation Institute,Nanjing University of Science and Technology,Nanjing 210094,China; 2.College of Biological,Chemical Sciences and Engineering,Jiaxing University,Jiaxing 314001,China; 3.The 42nd Institute of the Fourth Academy of CASC,Xiangyang 441003,China)
Molecular dynamics(MD)simulation was applied to propellant/liner model(PEG/NG/BTTN)/NPBA/HMX/AP/PEG/N-100//HTPB/TDI at 295 K with NVT ensemble.Component concentration distribution and migration behavior were presented and analyzed.The simulation results show that HMX and NPBA have a tendency to migrate to interface,while AP tends to have a homogeneous distribution.Moreover,a new model built by replacing HMX with RDX equivalently was studied by the molecular dynamic simulation.It is found that tensile modulus(E),bulk modulus(K),shear modulus(G)and Cauchy pressure(C12-C44)of the new formula decreas apparently.This implies that its stiffness,strength and ductibility are all decreased;The maximum bond length of the N—NO2trigger bonds of the new formula(1.528 Å) is larger than that of the original formula(1.503 Å),indicating that the sensitivity increases and safety decreases of the new formula.The results shows that the binding energies of HMX and other constituent is larger than RDX and other constituent indicate that compatibility of HMX and other constituent,which is better than RDX and other constituent.
propellant/liner interface;constituent migration;mechanical property;sensitivity;compatibility;molecular dynamics simulation
2013-07-31;
2014-01-16。
国防973项目(613142010102;613142010302)。
朱伟(1977—),男,博士/副教授,研究方向为计算化学及其应用。E-mail:zhuwmail@126.com
肖继军(1964—),男,博士生导师,主要从事高分子物理和分子材料学研究。E-mail:xiao_jijun@163.com
V512
A
1006-2793(2014)05-0678-06
10.7673/j.issn.1006-2793.2014.05.018
