新型二酰胺穴醚与吡啶N-氧化物的络合作用研究
2014-08-28赵淑婷蒋腊生
赵淑婷, 李 媚, 曾 志, 张 培, 唐 陆, 陈 坤, 蒋腊生
(华南师范大学化学与环境学院,广州 510006)
超分子化学研究分子如何通过非共价键相互作用自组装成分子聚集体[1].在超分子领域,主客体化合物分子之间通过弱相互作用(氢键、亲水-疏水作用、电荷转移作用、以及π-π堆积作用[2])或这些作用的累加从而实现分子识别、自组装等行为.冠醚、杯芳烃、环糊精、葫芦脲及其它大环化合物[3]作为常见的主体化合物,因易与中性或离子客体化合物形成主客体络合物,被广泛应用于合成机械互锁结构的超分子化合物[4].
近年来,以穴醚为主体研究其与不同客体化合物之间的相互作用越来越引起人们的关注.Gibson课题组[5]和黄飞鹤课题组[6]研究了不同结构及不同“空腔”大小的穴醚与paraquat及diquat的相互络合作用,利用非共价键相互作用合成了轮烷、索烃及可调控的分子机器[7];Fritz Vögtle等[8]设计合成了含有4个酰胺键的大环分子的亲核试剂,与溴代烃、酰氯等线性分子反应,利用氢键作用高产率合成了轮烷分子.另外,吡啶N-氧化物是一类重要要的有机化工中间体,广泛应用于医药、染料、催化等诸多化工领域[9-10].
最近,本课题组首次报道了二酰胺大环化合物与吡啶N-氧化物的相互作用.吡啶N-氧化物与通过氢键作用及空间匹配性进入二酰胺大环化合物中,形成1∶1稳定的准轮烷结构络合物,并可通过酸碱调控实现分子开关功能[11].基于本课题组所做的研究,设想把二酰胺引入穴醚中,设计并合成基于二苯并26-冠-8/二苯并32-冠-10的新型二酰胺穴醚5和10.目前,穴醚与吡啶N-氧化物(特别是双吡啶N-氧化物)的主客体络合物的研究较少.本文合成了新型二酰胺穴醚5和10,并探讨三维笼状穴醚与吡啶N-氧化物的络合作用及其结构与络合能力的关系.本文所涉及的主客体化合物的分子结构见图式1 (Scheme 1).

图式1 主客体化合物的结构图
1 实验部分
1.1 主要试剂和仪器
NMR采用美国varian NMR system 400 M 共振仪(400 MHz),溶剂为CDCl3, 纯度≥98%,TMS作内标;LR-MS 采用LCQ Advantage液质联用仪,ESI源;HRMS采用美国Bruker Apex IV FTMS,ESI源;熔点测定采用XT-4双目显微熔点测定仪,温度计未经校正;所用试剂均为市售A.R.级;柱层析硅胶为青岛海洋化工厂生产,未经活化;反应溶剂都经过无水处理.
1.2 实验步骤
1.2.1 化合物5的合成1-3参照文献[12]方法合成(图式2).1:产率24%, m.p.128.5~131.3 ℃(文献值:131.2~133.2 ℃);1H NMR (400 MHz, CDCl3, 22 ℃):δ=7.16 (d,J=4.0 Hz, 4H, Ar-H), 6.67 (s, 2 H, Ar-H), 4.11 (t,J=4.0 Hz, 8 H,α-OCH2), 3.87 (s, 6 H, -COOMe), 3.85 (t,J=4.0 Hz, 8 H,β-OCH2), 3.73 (s, 8 H,γ-OCH2);13C NMR (100 MHz, CDCl3, 22 ℃):δ=166.80, 159.80, 131.84, 107.95, 107.12, 71.02, 69.58, 67.75, 52.24.2:产率92%, m.p. 138~139.3 ℃(文献值: 139.0~140.0 ℃).1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.49(s, 4H, Ar-H), 6.36 (s, 2 H, Ar-H), 4.56 (s, 4 H, Ar-H), 4.05 (t,J=4.0 Hz, 8 H,α-OCH2), 3.84 (t,J=4.0 Hz, 8 H,β-OCH2), 3.73 (s, 8 H,γ-OCH2);13C NMR (100 MHz, CDCl3, 22 ℃):δ=160.15, 143.48, 105.42, 101.07, 71.01, 69.77, 67.54, 65.27.3:产率56%, m.p. 96~97 ℃;1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.46 (s, 6 H, Ar-H), 4.22 (s, 4 H, benzyl-H), 4.08 (t,J=4.0 Hz, 8 H,α-OCH2), 3.86~3.84 (m, 8 H,β-OCH2), 3.73 (s, 8 H,γ-OCH2).13C NMR (100 MHz, CDCl3, 22 ℃):δ=160.32, 137.50, 106.90, 101.61, 71.05, 69.68, 67.57, 54.93; MS:m/z=(100%)=576.49 (M+NH4)+.

图式2 化合物5和10的合成
4: 将化合物3(0.7 g, 1.2 mmol) 和P(Ph)3(0.9 g, 3.6 mmol)加入装有20 mL THF的单口瓶中, 常温反应过夜.TLC (V(EA)∶V(PE)=5∶3)跟踪反应进程.反应结束后,减压蒸馏除去溶剂,加入去离子水30 mL, 用2 mol/L的HCl溶液调节pH<1, 用乙醚萃取多次.取其水相并加入4 mol/L的NaOH溶液调节pH>12, 用二氯甲烷萃取,有机相用无水硫酸镁干燥,浓缩得到淡黄色固体4为 0.38 g, 产率62%, m.p. 141.5~143 ℃;1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.47 (s, 4 H, Ar-H), 6.37 (s, 2 H, Ar-H), 4.08 (t,J=4.0 Hz, 8 H,α-OCH2), 3.85 (t,J=4.0 Hz, 8 H, β-OCH2), 3.76 (s, 4 H, benzyl-H), 3.73 (s, 8 H, γ-OCH2);13C NMR (100 MHz, CDCl3, 22 ℃):δ=160.20, 145.88, 105.76, 100.16, 71.04, 69.76, 67.50, 46.76; MS:m/z(100%)=507.40 (M+H)+.
5:将Et3N (1 mL, 6 mmol), CH2Cl2(80 mL) 在氮气保护下分别加入到装有2只恒压滴液漏斗的二口瓶中.在其中1只恒压滴液漏斗中加入20 mL溶有(0.5 g, 1 mmol)的化合物4的CH2Cl2溶液,在另外一只恒压滴液漏斗中加入20 mL 溶有(0.26 g, 1 mmol)的5-叔丁基间苯二甲酰氯的CH2Cl2溶液(制备方法:将5-叔丁基-苯二甲酸与过量的二氯亚砜在100~110 ℃下回流反应3 h至溶液变澄清,减压条件下蒸出过量的二氯亚砜得到白色固体,化合物4与5-叔丁基-苯二甲酰氯的比例按1∶1.25算).冰浴条件下,同时缓慢滴加以上2种原料,滴毕,室温搅拌过夜,TLC (V(CH2Cl2)∶V(Acetone) =1∶1)跟踪反应进程.反应结束后,加入1 mol/L的HCl溶液调节pH至7,用CH2Cl2萃取3次,合并有机相,用无水硫酸镁干燥,浓缩得到粘稠的橙红色液体.柱色谱分离TLC [V(CH2Cl2)∶V(Acetone) =1∶1] 得到白色固体5为 0.40 g, 产率57%, m.p: 239.5~241 ℃;1H NMR (400 MHz, CDCl3, 22 ℃):δ8.22 (s, 2H, Ar-H), 7.62 (s, 1H, Ar-H), 6.73 (s, 2H, -CONH-), 6.46 (s, 6H, Ar-H), 4.44 (d,J=3.4Hz, 4H, benzyl-H), 4.08 (s, 8H, α-OCH2), 3.82 (s, 8H, β-OCH2), 3.69 (s, 8H, γ-OCH2), 1.39 (s, 9H, -CH3);13C NMR (100 MHz, CDCl322 ℃):δ=164.26, 159.95, 152.67, 139.02, 131.16, 127.94, 108.05, 100.90, 70.45, 69.66, 67.00, 46.05; MS:m/z(100%)=693.64(M+H)+.
1.2.2 化合物10的合成6-8参照文献[12]方法合成.6:产率32%, m.p: 105~106 ℃;1H NMR (400 MHz, CDCl3, 22 ℃):δ=7.15 (d,J=4.0 Hz, 4 H, Ar-H), 6.68 (d,J=4.0 Hz, 2 H, Ar-H), 4.10 (d,J=4.0 Hz, 8 H,α-OCH2), 3.87 (s, 6 H, -COOMe), 3.87~3.81 (m, 8 H, α-OCH2), 3.71 (d,J=4.0 Hz, 16 H,γ-OCH2,δ-OCH2);13C NMR (100 MHz, CDCl3, 22 ℃):δ=166.81, 159.79, 131.85, 108.05, 106.86, 70.97,70.91, 69.63, 67.89, 52.28. 7:产率32%, m.p:98~99.5 ℃,1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.49 (s, 4 H, Ar-H), 6.35 (s, 2 H, Ar-H), 4.54 (s, 4 H, benzyl-H), 4.14~3.94 (m, 8 H,α-OCH2), 3.90~3.75 (m, 8 H,β-OCH2), 3.69 (d,J=4.0 Hz, 16 H,γ-OCH2,δ-OCH2), 2.53 (s, 2 H, -OH);13C NMR (100 MHz, CDCl3, 22 ℃):δ=159.95, 143.71, 105.50, 100.74, 70.82, 70.77, 69.76, 67.55, 64.94. 8:产率56%, m.p:77.5~78.5 ℃;1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.45 (s, 6 H, Ar-H), 4.22 (s, 4 H, benzyl-H), 4.07 (s, 8 H, α-OCH2), 3.84 (s, 8 H, β-OCH2), 3.71 (d,J=4.0 Hz, 16 H, γ-OCH2,δ-OCH2);13C NMR (100 MHz, CDCl3, 22 ℃):δ=160.30, 137.53, 107.01, 101.29, 70.99,70.97, 69.73, 67.72, 54.89; MS:m/z(100%)=669.54 (M+Na)+.
9:方法同化合物4的合成,产物为浅黄色液体,产率56%,1H NMR (400 MHz, CDCl3, 22 ℃):δ=6.46 (s, 4 H, Ar-H), 6.36 (s, 2 H, Ar-H), 4.07~4.05 (m, 8 H,α-OCH2), 3.84~3.82 (m, 8 H,β-OCH2), 3.74 (s, 4 H, benzyl-H), 3.71 (d,J=8.0 Hz, 16 H,γ-OCH2,δ-OCH2).13C NMR (100 MHz, CDCl3, 22 ℃):δ=160.04, 145.59, 105.82, 99.80, 70.83, 69.69, 67.50, 46.50.
10:方法同化合物5的合成,得白色固体10, 产率,56%, m.p: 253.6~255.9 ℃.1H NMR (400 MHz, CDCl3, 22 ℃)δ8.20 (s, 2H, Ar-H), 7.40 (s, 1H, Ar-H), 6.50 (s, 4H, Ar-H), 6.41 (s, 2H, Ar-H), 6.34 (s, 2H,-CONH-), 4.49 (d,J=4.7 Hz, 4H, benzyl-H), 4.05 (s, 8H,α-OCH2), 3.79 (s, 8H,β-OCH2), 3.65 (d,J=3.9 Hz, 16H,γ-OCH2,δ-OCH2), 1.39 (s, 9,H- CH3).13C NMR (100 MHz, CDCl3, 22 ℃):δ=166.74, 160.05, 152.91, 139.79, 133.91, 128.84, 119.99, 107.63, 100.56,70.66,69.56, 67.46, 44.99, 35.15,31.17. MS:m/z=798.54 [10+NH4]+.
1.2.3 化合物GT3和GT4的合成 化合物GT3的合成[13]:将5 g 3,5 -二甲基吡啶(0.046 7 mol),40 mL冰醋酸放在装有回流装置的250 mL二口瓶中;升温至85 ℃, 开始滴加8 mLw(H2O2)=30%的双氧水;滴加完毕,搅拌3 h; 再滴加5.4 mL双氧水,搅拌22 h; 冷却至室温,减压蒸出蒸馏水-冰醋酸的混合溶液;用碳酸氢钠饱和水溶液中和至弱碱性,滤出固体;滤液用三氯甲烷萃取,然后用无水硫酸钠干燥有机相,过滤;减压蒸出溶剂,将得到的浅黄色固体用石油醚冲洗,得5.6 g白色固体,产品收率为97%. Mp:98~102 ℃ (文献值: 99~103 ℃),1H NMR (400 MHz, CDCl3)δ7.93 (s, 2H), 6.95 (s, 1H), 2.28 (s, 6H).
化合物GT4的合成[14]:将2.5 g 4,4-二甲基-2,2联吡啶(13.57 mmol),18 mL冰醋酸放在装有回流装置的100 mL双口瓶中;升温至85 ℃,开始滴加3.5 mLw(H2O2)=30%的双氧水;滴加完毕,70~80 ℃反应4 h;再滴加2 mL双氧水,反应过夜;冷却至室温,浓缩除去过量的水和乙酸, 然后将溶液倒入约200 mL丙酮中,有白色沉淀产生,抽滤,用丙酮和乙醚洗涤,真空干燥后得白色固体2.05 g, 产品收率为为71%.1HNMR (400 MHz)δ8.23 (d,J=5.3 Hz, 2H), 8.23 (d,J=5.3 Hz, 2H), 7.49 (s, 2H), 7.14 (s, 2H), 2.38 (s, 6H).
1.2.4 ESI质谱分析 通过电喷雾质谱技术分别测定分子5和GT3、GT4以及10和GT3、GT4在乙腈中的混合溶液质谱,检测两者形成络合物的准分子离子峰,确定准轮烷结构中主客体分子的化学计量比.
1.2.5 核磁滴定实验 络合常数常用作表征主客体间形成络合物的稳定性,也是用来判断超分子体系形成能力的最主要参数之.本文采用1H NMR滴定法测定络合常数.核磁共振滴定实验使用美国varian NMR system 400 M 核磁共振仪在295 K测试,将主体和客体化合物分别用CDCl3溶解.核磁滴定法测定中,固定穴醚主体的浓度为1.0×10-3mol/L, 用微量进样器将客体溶液(100×10-3mol/L)逐次滴加到定量主体溶液中,分别在仪器恒温295 K 时测定1H NMR, 分别记录主体苯环上的H6在不同条件下的化学位移值.由于主体与客体之间形成超分子络合物时引起主体中质子的化学位移的变化,基于这种化学位移的变化,用Benesi-Hildebrand method方法计算出主客体相互络合的络合常数[14].用记录下的每次滴加客体时,主体化合物5和化合物10苯环上质子H6的化学位移变化值Δδ的倒数为纵坐标,以不断增加的客体GT3、GT4浓度的倒数为横坐标作图得到Benesi-Hildebrand plots.
2 结果与讨论
2.1 二酰胺穴醚与吡啶N-氧化物的络合作用
2.1.1 电喷雾质谱(ESI-MS)分析 在主客体化学研究中,电喷雾质谱技术(ESI-MS)是检测主客体分子形成络合物的常用方法之一.本文运用该技术分别测定了吡啶N-氧化物GT3和GT4与穴醚5和10混合的乙腈溶液, 通过对MS 图进行分析确定了络合物中主客体分子的化学计量比.以5与GT3的ESI-MS图为例,图1为5与GT3于乙腈中的ESI-MS 图.基峰m/z=247.03(100%),对应于[GT32+H]+, 即GT3二聚的准分子离子峰;另外,图中有另一组峰m/z=816.35(40%),对应于[5·GT3+H]+,即主客体化合物以化学计量比1∶1形成络合物的准分子离子峰,除此之外图中并无明显的其它络合物的峰.因此可判定该络合物结构中主客体化合物化学计量比为1∶1.
5与GT4的ESI-MS 图中:m/z=931.28(100%),对应于[5·GT4+Na]+;10与GT3的ESI-MS 图中,m/z=904.34(45%),对应于[10·GT3+H]+;10与GT4的ESI-MS 图中,m/z=1 019.07 (100%),对应于[10·GT4+Na]+.
综上可知:ESI-MS 结果初步证明穴醚5和10能与吡啶N-氧化物GT3、GT4形成1∶1 的络合物,主客体之间存在较强的相互作用,其络合物体系在实验测定条件下具有较高的稳定性.

图1 化合物5与GT3的混合溶液的低分辨质谱图, m/z 816.35 [5·GT3+H]+, 247.03 [GT32 +H]+
2.1.2 络合物体系的1H NMR分析 客体化合物GT3、GT4是极性较强的化合物,难溶于弱极性的有机溶剂(如:CH2Cl2及乙醚),但在CDCl3中有较好的溶解性.综合主客体的溶解性,本文采用CDCl3为溶剂溶解主客体化合物并在恒温295 K时进行1H NMR测试.首先,利用1H NMR图谱监测等摩尔比的主体化合物5和10与客体化合物GT3、GT4形成络合物的过程.以等摩尔比5与GT3相互作用为例,图2为等摩尔比5与GT3混合溶液的部分1H NMR图(295 K).
等摩尔的主体5和客体GT3混合后,溶液颜色无明显变化,但是客体吡啶环上的质子Ha和主体苯环上的质子(H6和H7)都明显地向高场移动,这是苯环的屏蔽效应所致.主体苯环上的质子H6变化最为显著,这表明在客体GT3的吡啶环和主体化合物5中2个苯环形成了强的 π-π 堆积作用.主体穴醚5中叔丁基苯环上的质子H3和酰胺上的质子H4都明显地向低场移动(去屏蔽效应所致).通过以上的化学位移变化可初步判定,客体GT3进入穴醚5的“空腔”中形成络合物.结合5与GT3的ESI-MS图和等摩尔的1H NMR图,推测5与GT3可能形成的络合物的结构图如图式3.

图2 (a)5, (b) 等摩尔混合的5与GT3, (c)GT3的部分核磁图 (8.0 mmol/L, CDCl3, 400 MHz, 295 K)
Figure 2 Partial1H NMR spectra of host5(a), equimolar mixture of5andGT3(b), and guestGT3(8.0 mM, CDCl3, 400 MHz, 295 K, c)
2.1.3 络合常数测定-1H NMR 滴定[16-17]为了进一步探讨形成络合物的稳定性,采用核磁滴定的方法测定了主客体化合物形成络合物的络合常数.该实验用CDCl3溶解样品,恒温295 K, 分别测得了5·GT3、5·GT4、10·GT3、10·GT4四组络合物的平衡常数.通过ESI-MS分析可知,主客体化合物之间均以化学计量比1∶1形成络合物,参照文献[15], 使用Benesi-Hildebrand公式对核磁滴定数据进行处理计算络合物的平衡常数.以5·GT3络合常数为例,固定5的浓度(1.0 mmol/L),逐次加入的等浓度GT3溶液分别测试,得到一系列不同客体浓度的1H NMR图,用记录下的每次滴加时主体化合物5苯环上的质子H6的化学位移变化值Δδ的倒数为纵坐标,以不断增加的客体GT3浓度的倒数为横坐标作图得到Benesi-Hildebrand plots图,如图3中 (a) 所示:R2=0.996 1,说明实验所得数据点能很好的拟合成一条直线,该直线的截距intercept=5.501,斜率slope=90.40,K=intercept/slope×103.然后通过误差传递公式[18]计算实验的测量误差,最终得到主体化合物5和客体化合物GT3形成的络合物的平衡常数K=60.85±1.35.

图式3 化合物5与GT3形成络合物的结构
图3为通过核磁滴定实验测定络合物5·GT3、5·GT4、10·GT3、10·GT4络合常数绘制的Benesi-Hildebrand线形图,由直线斜率计算可得的络合常数分别为60.84±1.35, 63.61±1.60, 1 449.22±43.82, 1 547.33±64.61(表1).

表1 不同络合物的络合常数(CDCl3,295 K)Table 1 Association constants for the complex in CDCl3 at 295 K
从表1中的数据可知,K10·GT4>K10·GT3>K5·GT4>K5·GT3. 从K5·GT4>K5·GT3,K10·GT4>K10·GT3来看,对于相同的主体化合物,双吡啶氮氧化物与单吡啶氮氧的络合能力相差不大.可能的原因是,双吡啶N-氧化物存在空间异构,未能与酰胺键上的N原子形成更多的氢键.而K10·GT4>K5·GT4,K10·GT3>K5·GT3, 对于相同的客体,穴醚10与不同客体的络合能力都比穴醚5与相应客体的络合能力大,且化合物10的络合常数大约是化合物5络合常数的24倍.说明了空腔大小对主体和与吡啶N-氧化物的络合能力的影响起主要作用[6,19].其原因可能是:客体的尺寸较大,未能完全进入穴醚5的空腔,形成的 π-π 堆积作用和氢键作用较弱;而穴醚10具有更大的环状空腔,客体完全进入空腔中,形成较强的 π-π 堆积作用和氢键作用,络合能力更强.
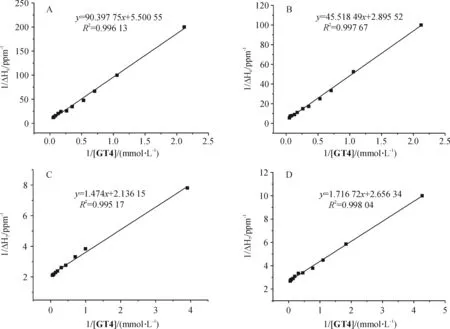
图3 (A)化合物5与GT3, (B) 化合物5与GT4, (C) 化合物10与GT3, (D) 化合物10与GT4的Benesi-Hildebrand关系图
Figure 3 Benesi-Hildebrand plots for the formation of complex, molecule5withGT3(A), molecule5withGT4(B), molecule10withGT3(C), and molecule10withGT4(D) based on the data for proton H6at 295 K in CDCl3, [H]o=1.0 mmol/L
3 结论
本文设计并合成了具有空腔大小不同并具有多重识别位点的二酰胺穴醚5和10,并对化合物进行了结构表征.采用ESI-MS和NMR技术对主体二酰胺穴醚5和10与客体吡啶N-氧化物的络合作用进行了对比研究.实验结果表明:在CDCl3中2种穴醚主体均能与吡啶N-氧化物客体形成1∶1的络合物;络合常数测定结果进一步表明,客体的不同对络合常数的影响不明显,而主体化合物“空腔”大小不同对络合常数的影响非常显著.“空腔”较大的主体10的络合能力是比“空腔”相对较小的主体5的络合能力大23倍.由此证明, 设计合理的空间匹配的主客体化合物,对提高主客体的络合能力,为进一步合成机械互锁型分子提供重要的参考意义.
参考文献:
[1] Fouquey C, Lehn J, Mlevelut A M. Molecular recognition directed self-assembly of supramolecular liquid crystalline polymers from complementary chiral components[J]. Advanced Materials, 1990, 2:254-257.
[2] Lehn J M. Upramolecular chemistry-scope and perspectives molecules, supermolecules, and molecular devices (nobel lecture)[J]. Angewandte Chemie International Edition, 1988, 27(1):89-112.
[3] Raymo F M, Stoddart J F. Interlocked macromolecules[J]. Chemical Reviews, 1999, 99:1643.
[4] 周其忠, 何春林, 黄飞鹤, 等. 机械互锁结构分子的模板合成[J]. 化学通报, 2008, 71:809-815.
Zhou Q Z, He C L, Huang F H,et al. Templated syntheses of molecules with mechanically interlocked structures[J]. Chemistry, 2008, 71:809-815.
[5] Zhang J Q, Huang F H, Gibson, H W. Paraquat substituent effect on complexation with a dibenzo-24-crown-8-based cryptand[J]. The Journal of Organic Chemistry, 2007, 72:8935-8938.
[6] Zheng B, Wang F, Huang F H, et al. Supramolecular polymers constructed by crown ether-based molecular recognition[J]. Chemical Society Reviews, 2012, 41:1621-1636.
[7] Barrell M J, Leigh D A, Lusby P J. An ion-pair template for rotaxane formation and its exploitation in an orthogonal interaction anion-switchable molecular shuttle[J]. Angewandte Chemie International Edition, 2008, 47: 8036-8039.
[8] Hubner G M, Glaser J, Vogtle F, et al. High- yielding rotaxane synthesis with an anion template[J]. Angewandte Chemie International Edition, 1999, 38:383-386.
[9] Chen J, Takenaka N. Helical chiral pyridine N-oxides:A new family of asymmetric catalysts[J]. Chemistry-A European Journal, 2009, 15:7268-7276.
[10] 蔡雪萍,许旋,甘桂莲,等.ITO电极上Tris促进[Ru(bpy)3]2+对次黄嘌呤的电催化氧化[J].华南师范大学学报:自然科学版,2013,45(5):68-73.
Cai X P, Xu X, Gan G L, et al. Tris-enhanced electrocatalytic oxidation of hypoxanthine by [Ru(bpy)3]2+on an ITO electrode[J]. Journal of South China Normal University: Natural Science Edition, 2013, 45(5): 68-73.
[11] Chen M J, Han S J, Jiang L S, et al. New switchable [2]pseudorotaxanes formed by pyridine N-oxide derivatives with diamide-based macrocycles[J]. Chemical Communications, 2010, 46:3932-3934.
[12] Xu Z K, Jiang L S, Feng Y H, et al. One-pot synthesis of donor-acceptor [2]rotaxanes based on cryptand-paraquat recognition motif[J]. Organic & Biomolecular Chemistry, 2011, 9:1237-1243.
[13] 徐宝财, 王关兴, 辛秀兰, 等. 2, 3-二甲基-N-氧化吡啶的合成[J]. 精细化工, 2007, 24(7):681-683.
Xu B C, Wang G X, Xin X L, et al. Synthesis of 2, 3-Dimethylpyridine-N-oxide[J]. Fine Chemicals, 2007, 24(7):681-683.
[14] Zhou W Z, Feng X J, Ke H S. New polyoxometalate-based mononuclear lanthanide complexes with slow relaxation of magnetization[J]. Inorganica Chimica Acta, 2012, 394:770-775.
[15] Benesi H A, Hildebrand J H. The benesi-hildebrand method for determination of Kffor DA association and ε values for DA-CT absorption[J]. Journal of the American Chemical Society, 1949, 71:2703-2705.
[16] Tomimasu N, Kanaya A, Harada A. Social self-sorting:Alternating supramolecular oligomer consisting of isomers[J]. Journal of the American Chemical Society, 2009, 131:12339-12343.
[17] Huang F H, Gibson H W. A supramolecular poly[3]pseudorotaxane by self-assembly of a homoditopic cylindrical bis(crown ether) host and a bisparaquat derivative[J]. Chemical Communications, 2005, 13:1696-1698.
[18] 杨登科, 陈木娟, 蒋腊生, 等. 二酰胺大环化合物与吡啶N-氧化物的络合作用研究[J]. 化学学报,2012, 70(12):1385-1393.
Yang D K, Chen M J, Jiang L S, et al. Studies on the complexation of di-amide based macrocycles with pyridine N-oxides[J]. Acta Chimica Sinica,2012,70(12):1385-1393.
[19] Yan X Z, Wei P F, Huang F H, et al. [2]Pseudorotaxanes based on the recognition of cryptands to vinylogous viologens[J]. Organic Letters, 2011, 13(24): 6370-6373.
