HPLC法测定肠虫清胶囊中阿苯达唑的含量
2014-08-23游国叶熊大伟
游国叶,熊大伟
(信阳职业技术学院 药学院,河南 信阳 464000)
肠虫清胶囊的主药成分为阿苯达唑,阿苯达唑(Albendazole,ABZ)又名丙硫咪唑,化学名为5-丙硫基-1H-苯并咪唑-2-氨基甲酸甲酯,为一广谱高效低毒的驱虫药.该药由美国Smith Kline公司1977年首次上市,临床上广泛用于治疗各种蠕虫感染性疾病,其作用机理主要在于抑制虫体延胡索酸还原酶,阻止虫体能量的生成,从而达到杀灭虫体的目的。该药对人体及动物寄生的线虫、吸虫、涤虫均有强大的驱杀作用,并可显著抑制虫卵发育,在体内无积蓄作用,其驱虫谱、疗效及毒性等均优于目前已投产的其他抗蠕虫药物,该药现已被列入医保产品,成为临床上用于治疗各种蠕虫感染性疾病的首选药物[1]。在阿苯哒唑原料的现行标准中,其含量测定在美国药典29版、欧洲药典5.0版和中国药典2010年版[2]中均为滴定法,其胶囊剂含量测定在中国药典2010年版规定的方法为分光光度法,利用295nm处的吸收系数计算含量[2]。HPLC法测定阿苯达唑的含量的报道较为少见[3],我们通过建立HPLC法测定肠虫清胶囊中阿苯达唑的含量,方法快速、准确,重现性好。
1 仪器与试药
1.1 仪器
Waters2695高效液相色谱仪,Waters2996紫外检测器 (美国Waters公司);BP211D型半微量电子天平;PHS—2CA数字式酸度计(上海理达仪器厂)。
1.2 试药
肠虫清胶囊 (新乡市同心药业,批号20080110,20080113,20080117,规格0.2g);阿苯达唑对照品 (中国药品生物制品检验所,批号100373-200301);纯化水(新乡市平安药业有限公司);甲醇为色谱纯(江苏国达,200704),其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
阿苯达唑为碱性化合物,和硅胶柱的硅醇基有较强的相互作用,会造成强保留,因此应选择硅醇相互作用弱的色谱柱,Luna C18的硅醇相互作用约为0.2[3],能够获得理想的分析结果。
流动相之一应该选择缓冲液,以获得稳定的保留时间,同时为了解决阿苯达唑的溶解性问题,选取0.1 mol/L醋酸铵缓冲液(用醋酸调节pH至3.1)。当缓冲液和甲醇的比例调整到40∶60时,阿苯达唑的保留时间在稳定在9.8 min左右。流量为1 mL/min,柱温20℃。
取阿苯哒唑对照品用醋酸溶解,流动相稀释成1 mg/mL,在200~400 nm的波长范围内扫描,阿苯哒唑在288 nm处有最大吸收,故紫外检测器的波长为288 nm。
所以色谱条件为:
色谱柱:Luna C18(5μm,(250×4.6)mm);流动相:甲醇-0.1 mol/L醋酸铵缓冲液(用醋酸调节pH至3.1)(60:40);检测波长:288 nm;流量:1 mL/min;进样量:20 μL;柱温:20℃。
在上述色谱条件下,阿苯达唑的色谱图见图1,和杂质峰的分离度大于1.5,拖尾因子1.03,理论塔板数按阿苯达唑计算大于3 500。

图1 阿苯达唑的色谱图
2.2 溶液制备
2.2.1 对照品溶液的制备 精密称取阿苯达唑对照品50 mg,置50 mL量瓶中,加醋酸10 mL,振摇使溶解,加甲醇稀释至刻度,摇匀,准确量取5.0 mL置10 mL量瓶中,加流动相定容,摇匀,稀释成0.5 mg/mL对照品液。
2.2.2 供试品溶液的制备
取本品内容物,研细,精密称取适量(约相当于阿苯达唑50 mg),置50 mL量瓶中,加醋酸10 mL,振摇使溶解,加甲醇稀释至刻度,摇匀,滤过,取续滤液5.0 mL置10 mL量瓶中,加流动相定容,摇匀,即得。
2.3 线性关系考察
精密称取干燥至恒重的阿苯达唑对照品50 mg,置50 mL量瓶中,加醋酸10 mL,振摇使溶解,加甲醇稀释至刻度,摇匀,即得阿苯达唑对照品储备液。分别准确量取1.0,2.0,4.0,5.0,6.0,8.0和10.0 mL阿苯达唑对照品储备液置10 mL量瓶中,加流动相定容,摇均,即成0.1,0.2,0.4,0.5,0.6,0.8,1.0 mg/mL系列溶液,按2.1项下色谱条件测定峰面积,以峰面积对浓度作线性回归,结果见图2。结果表明,阿苯达唑在0.1~1.0 mg/mL浓度范围内A=2.85×106C+6.5×104,r=0.999 5,线性关系良好。

图2 阿苯达唑的线性关系
2.4 精密度试验
取0.5 mg/mL阿苯达唑对照品溶液,按2.1项下色谱条件进样6次,测定峰面积,精密度试验RSD为0.40%,精密度良好。
2.5 重复性试验
取同一批号20080113样品,按2.3项下方法制备6份供试液,按2.1项下色谱条件进样,测定峰面积,重复性试验RSD为0.66%,重复性良好。
2.6 加样回收率试验
精密称取已知含量的样品(20080113)细粉约50 mg,分别加入高、中、低(20 mg、40 mg、60 mg)不同量干燥至恒重的阿苯达唑对照品,各3份,按含量测定方法进行测定,计算回收率,结果见表1。

表1 回收率试验结果
2.7 稳定性试验
取同一供试品溶液在放置时间为0、2、4、6、10 h时进样,测定峰面积,稳定性试验RSD为0.52%,稳定性良好。
2.8 定量限
按信噪比为3(S/N=3)对最低检测限进行测定,结果表明,当进样浓度为0.25μg/mL时,阿苯哒唑峰信号约为基线噪音的3倍,即最低检测浓度为0.25μg/mL。
3 样品含量测定
取3批样品各20粒胶囊,倾出内容物,囊壳用小刷拭净,精密称定,精密称取适量(约相当于阿苯达唑50 mg),按2.2.2项下方法制备供品溶液。分别取供试液和2.2.1项下对照品液,在2.1项下色谱条件进样,用峰面积外标法计算含量,结果见表2。

表2 样品含量测定结果
注:样品含量计算方法为
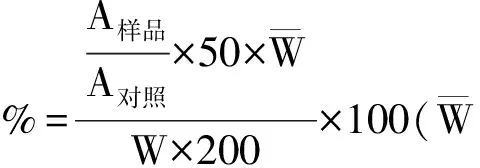
4 讨论
阿苯达唑微溶于丙酮、乙酸,不溶于乙醇、水,溶于乙酸[3],制约了流动相的选择,采用甲醇-醋酸铵缓冲液为流动相,通过调整流动相比例和PH值,既解决了溶解性问题,也获得了较好的系统适用性。
虽然药典规定的分光光度法在295 nm波长处测定吸收值,但在我们实验的色谱条件下,阿苯达唑的最大吸收在288 nm处,这是因为在pH3.1时,阿苯达唑以离子状态存在,共轭结构减少,最大吸收向短波方向移动。
与中国药典规定的分光光度法相比,本法能够排除杂质干扰,准确度高,重复性、稳定性好。
参考文献:
[1] 万启惠.广谱驱蠕虫新药—— 丙硫咪唑[J].遵义医学院学报,1988,11(1):73-74.
[2] 国家药典委员会.中华人民共和国药典(二部)[M].北京:化学工业出版社,2005:290.
[3] 邵超.HPLC法测定阿苯达唑颗粒的含量[J].黑龙江医药,2011,3(5):31.
