高脂血症长爪沙鼠模型的转录组检测和代谢性炎症通路的初步研究
2014-08-14刘月环毛栋森吴旧生钟宇森周莎桑金晓音柯贤福应华忠
刘月环,毛栋森,吴旧生,钟宇森,周莎桑,金晓音,柯贤福,应华忠
(1.浙江省医学科学院,杭州 310013;2.浙江大学动物科学学院,杭州 310058)
高脂血症是由脂肪代谢或运转异常使血浆中一种或几种脂质高于正常的症状。流行病学调查表明,高脂血症是诱发动脉粥样硬化、冠心病、高血压、糖尿病、胰腺炎、癌症等多种疾病的主要危险因素之一[1]。2002年全国第四次营养调查研究覆盖了全国31个省市,根据调查的数据估计,全国血脂异常的人群约1.6亿[2],由此可见,探索发病机理,研究高脂血症防治措施和筛选有效治疗药物是今后的一项长期工作。理想动物模型是研究工作获得重要进展的关键之一[3],目前国内实验室使用较多的动物是大、小鼠,已有许多证据表明,大鼠和小鼠血清脂蛋白构成、分布和体内脂质代谢过程与人类存在较大差距,不能真实反映人的脂代谢状态,且对高胆固醇不敏感。猴、猪、狗等大型动物病变与人类相似,但成本高、来源困难、造模时间较长。兔是胆固醇敏感动物,但其血清高密度脂蛋白比例过高,形成病变时间较长等,其使用受到限制。转基因动物模型为血脂代谢和药物评价提供了全新的实验体系,但人类脂代谢紊乱和动脉粥样硬化的形成是多基因所致,很难归咎于单基因异常,同时由于传代后遗传性状的稳定性,价格昂贵等系列原因使其未能成为常用的高脂血症和动脉粥样硬化动物模型[4]。
自上个世纪六十年代以来,国内外学者以高脂饲料诱发的长爪沙鼠高脂血症的研究,结果表明该动物以其成模时间短(只需要四周)、性状典型(与人类高脂血症极为相似)、饲料配方简便(不需要甲状腺抑制药物)而著称[5-6],但其遗传基础一直不明。为查明该问题,筛选培育高脂血症长爪沙鼠新品系的遗传标记,笔者自2005年以来建立了长爪沙鼠高脂饮食的高脂血症模型[7],用候选基因法对APOE、LCAT两个基因的遗传多态性进行了研究,发现在Z:ZCLA封闭群长爪沙鼠大群体中LCAT呈单态,而在ApoE基因发现了三个SNP,用这三个SNP对ZCLA两个微生物等级封闭群进行了相应的遗传结构及遗传效应的分析评价后,认为这三个SNP对于封闭繁育了30多年的ZCLA群体来说标记偏少,基因频率偏低,于是我们引入新兴的转录组技术,从基因转录水平出发,阐明与高脂血症相关的关键基因的转录状态及基因转录及表达的调控网络,同时在群体水平上对该基因的遗传力进行评价,期望尽快找到可利用的主效基因,防止ZCLA群体优良基因的进一步丢失,加快筛选遗传标记的进程。
1 材料和方法
1.1 实验动物与试剂
实验用长爪沙鼠由浙江省医学科学院实验动物中心提供并饲养(实验动物使用许可证:SCXK(浙)2008-0034,SYXK(浙)2008-0114,商品化饲料参照GB14923-2010生产)。配合高脂饲料,选取90日龄长爪沙鼠(雌、雄性鼠兼用)60只,分为正常组(n=30)及模型组(n=30),体重(50~70)g,正常组饲喂基础饲料,而模型组则饲喂基础料70.5%(各成份的配比及生产按照GB14924-2001执行)、猪油7%、胆固醇2%、胆盐0.5%、蛋黄粉7%。造模型四周时取血清测定甘油三脂(TG)、总胆固醇(CHO/TC)、低密度脂蛋白(LDL/LIP)、高密度脂蛋白(HDL),饲养实验进行四周,处死前进行CO2麻醉,大体解剖观察肝脏形态,有明显脂沉积者取作转录组RNA制备的样本,两组动物分别组成两个RNA样本池。
RNA-Seq 测序cDNA 文库制备采用Illumina Satandard Kit 及QIA quick PCR purification KIT 试剂盒(Qiagen)试剂盒,具体操作按照说明书进行,测序在浙江大学纳米研究院(Illumina GA IIx 测序平台)。
1.2 RNA-Seq文库的制备,read质量的预处理及Unigene的de novo拼接
按照说明书,用Poly(T)寡聚核苷酸从上述2个总RNA池(正常组与模型组,每池取20 μg)中抽取带 poly(A)尾的RNA,70℃温度下裂解5 min,将其随机打断成片段。利用N6随机引物和反转录酶将片段化的mRNA 合成cDNA 一链,继而合成双链cDNA,然后对二链cDNA 进行末端修饰将其连接到Illumina 双端测序接头上(adapter),用于测序的cDNA 再经过15个循环的PCR 线性扩增后经富集和纯化得到最终的cDNA 文库。利用 通过Solexa RNA的paired-end测序进行5’ 和3’ 双向RNA-Seq 测序,每个泳道产生数百万条Read(样本数据)。鉴于Solexa数据错误率对结果的影响,对原始数据进行质量预处理(即用滑动窗口法去除低质量片段,窗口长度为5 bp,长度阈值35 bp,质量阈值20(错误率=1%)大小。将长爪沙鼠2组样本的测序reads合并进行de novo拼接,使用软件velvet_1.0.19,paired-end的拼接方法,得到许多unigene。
1.3 与公共数据库的BLAST及基因的KEGG注释与GO注释
将拼接样本unigene与公共数据gene进行比较,通过gene的同源性进行功能注释。基因相似比对主要是使用基因Basic Local Alignment Search Tool (BLAST)算法[8]。样本基因序列,分别与SWISS-PROT、CDD、PFAM、NR和TREMBL库进行比对,取相似度>30%,且e<1e-5的注释。利用WEGO对得到的基因进行gene ontology ( GO )分类[9-10],统计基因在Biological Process, Cellular Component, Molecular Function 三个类别的各GO term。进行KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway分析[11],此分析是基于预测得到ORF序列,利用KAAS预测得到对应的KO号,然后利用KO号对应到KEGG pathway上,分析基因与KEGG中酶注释的关系文件以及映射到pathway的信息。
1.4 计算长爪沙鼠基因表达丰度
用拼接得到的47 522个沙鼠基因做库,用序列相似性比对的方法求各基因在各样本中的表达丰度。使用软件bowtie 0.12.7,single-end的mapping方法,允许一个reads比对到多个基因上(-v 3-a—phred64-quals)。基因表达估计方法用RPKM来表示,即以每个基因单位长度序列数的RPKM 值(reads per kilobase of exon model per million mapped reads)来衡量,就是每百万读段中来自于某基因外显子每千碱基长度的读段数,公式如下:
1.5 差异表达基因分析及其GO和KEGG富集分析
应用DEGseq 程序包比较2个文库中差异基因表达的情况[12],根据各样本基因的表达丰度值(FPKM)做基因的差异表达分析,包括:fold change分析,fisher检验,chisq检验等差异表达分析。对于以上各方法得到的差异基因,以fold change结果为准。将差异基因作为前景基因,全部基因作为背景基因,进行GO和KEGG的富集分析,使用超几何分布算法(phyper)计算前景基因同GO/Pathway分类中某个特定分支的P值,并用FDR进行校正。
1.6 Q-PCR的验证
提取纯化的总RNA,检测28 s 和18 s 质量合格后,挑选10个差异倍数大于2的差异基因,设计引物,引物在上海睿迪生物公司合成。各目的基因引物序列以及退火温度见表1。用普通PCR 方法对每个目的基因和持家基因进行扩增,然后通过2%琼脂糖凝胶电泳分离,以确定每个基因扩增的特异性。在每板反应结束后,所有引物扩增系列均拟制溶解曲线,每个溶解曲线均只存在一个峰值,表明荧光定量PCR 扩增过程特异性较好,无非特异性扩增,这和前面普通PCR扩增电泳结果一致。实时定量PCR 结果采用2-ΔΔCt方法来表示。试验数据采用SPSS 13.0软件(SPSS Inc. Chicago, IL, USA)进行统计。采用配对T 检验法(Paired-Samples T Test)分析高脂血症对长爪沙鼠基因表达的影响。试验结果所有数据表示方式为:平均值±标准误(Mean±S.E)。P<0.05 表示差异显著,P<0.01表示差异极显著。

表1 转录组测序的Q-PCR引物
2 结果
2.1 测序数据的注释
RNA-seq数据共6.68千万条,平均长度94.63 bp,测序样本均base满足2G要求,De novo拼接后最终得到了有效鼠基因47 522个(即≥100 bp的unigene),大小26.9 Mb;约82.53%序列为比对到基因组上外显子的序列; 其中长于1 000 bp的unigene有8 015个(表2)。

表2 拼接结果
2.2 基因表达差异及显著性的分析
对得到的基因注释分析是基于BLAST UniProt的结果 ( 即合并与Swiss-Prot和trEMBL的结果 ),Blast能够实现比较两段核酸或者蛋白序列之间的同源性的功能,它能够快速的找到两段序列之间的同源序列并对比对区域进行打分以确定同源性的高低。利用得到的uniprot号比对GO term,拼接基因得到的所有注释,详细信息注释上NR、SWISS-PROT、CDD、PFAM、TREMBL库的基因分别有51.43%、49.20%、 50.93%、 32.65%、44.19%。
在level2的基础将5 365个GO分别按照分子功能(molecular function, MF)、生物过程(biological process, BP)和细胞成分(cellular component, CC)进行分类,以上三种方法的分类信息分别含有 621个GO (13.2%)、1 989个GO (37.1%)和706个GO (11.6%),剩下的为没有任何分类信息的基因。GO主要是信号传导、细胞粘附、细胞凋亡、细胞分化、免疫反应、炎症反应、氨基酸代谢、维生素合成与代谢,能量代谢、胆固醇代谢过程、脂肪酸代谢过程、脂质转运、细胞脂质代谢过程、甘油三酯代谢过程、脂质分解过程、脂肪细胞分化、脂肪酸分解过程、甘油三酯生物合成过程、细胞因子调节信号通路、胰岛素受体信号通路、胆固醇平衡、细胞因子形成等 (P<0.001)。 共有12 914个基因注释到282个pathway,其中包括1 004个酶,表3列示了注释上基因最多的5个pathway,图1(见彩插4)列示了长爪沙鼠unigene注释到ko00010(其中红色表示注释上的基因)。

表3 长爪沙鼠unigene注释上基因最多的5个pathway
2.4 Q-PCR验证
以肝脏组织cDNA 作为模板,对模板进行 10倍浓度梯度稀释,以优化后的反应条件进行扩增,得到各基因的标准曲线,标准曲线的R2值都大于0.980,且各基因的扩增效率都处于95%~105% 之间,目的基因与内参基因的扩增效率都接近100%,且它们之间扩增效率相对偏差不超过5%,可以用于荧光定量PCR分析(图2)。
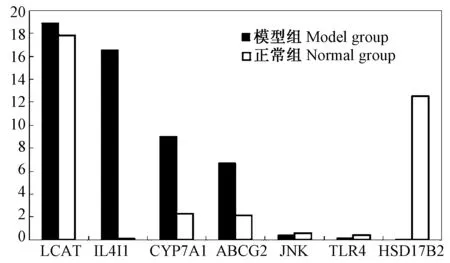
图2 七个基因Q-PCR验证的结果
使用2倍上调/下调的fold change阈值统计发现,本研究中,诱发高脂血症与正常动物之间共获得21 125 差异基因,16 087个下调,5 038个上调,总体上模型组相对于正常组存在较显著的趋势性下调关系。
2.5 与炎症通路相关的基因及其表达量
共有8个通路[13]与长爪沙鼠高脂血症代谢性炎症密切相关(P<0.01),分别是ECM受体的相互作用途径(ko04512)、细胞粘附分子途径 (ko04514)、细胞因子受体相互作用 (ko04060)、粘着 (ko04514/04510)、白细胞跨内皮迁移 (ko04670)、趋化因子信号转导通路 (ko04062)、补体和凝血级联 (ko04610)与抗原处理与演示 (ko04612),其余通路(如JAK-STAT信号通路 (ko04630)、MAPK信号通路(ko04010)、Toll样受体信号通路(ko04620)、B细胞受体信号通路(ko04662)、急性髓细胞白血病(ko05221)、溶酶体(ko04142),ABC转运子(ko02010)等均未达显著水平。对照组是实验组2倍以上的基因有LAMA1(层粘连蛋白)、LAMA2(层粘连蛋白)、ITGB3(整合,β3(Ⅲa的血小板膜糖蛋白CD61抗原))、MHC I (主要组织相容性复合体)、MHC II(主要组织相容性复合体)、CLDN5(紧密连接5)、PTPRC(蛋白络氨酸磷酸酶,受体类型C)、CD58(CD58分子)、PTPRC(细 胞 粘 附 分 子 途 径)、EPOR(细胞因子细胞因子受体相互作用途径)、PPPICB(蛋白磷酸酶1催化亚基,β同工酶)、ACTB(肌动蛋白,β)、JUN(c-Jun蛋白),XCR1(趋化因子(C模式)受体1)和CR1(补体受体I型)。15个与免疫和炎症相关的基因表达均显著下调,其中JUN表达下调五倍,EPOR(促红细胞生长素受体),XCR1(趋化因子(C模式)受体1),CR1(补体受体1型)下调超过3倍。
3 讨论
转录水平的调控是生物体最主要的调控方式,而RNA-Seq 技术最基本的应用也是检测基因的表达水平,它对同一样品深度测序可以捕获低表达的基因,而对大量样品同时测序可以获得样品之间的表达差异[14]。Mortazavi等[15]利用Solexa 技术进行了小鼠的4种不同组织的转录组测序,他们发现有90%的序列比对上基因组上的外显子区域,剩下的10%序列比对上的区域可能是未知的一些转录区域。Pan 等[16]利用Solexa 测序仪进行了人的转录组测序,首次利用新一代测序数据发现和检测了选择性剪切。对于高脂血症这样的多基因疾病,弄清疾病发作期病变器官(如肝脏)高表达的基因,全面地评价模型,或筛选出高脂血症的主基因或高效应SNP标记,培育高脂血症的新品系,具有十分重要而现实的科学意义。
代谢性炎症是由于摄入营养物和代谢过剩而触发炎症的过程,是一种低程度的系统性炎症。在长期的进化过程中,各种生物都形成了代谢和免疫反应的公共通路,也可以说机体对营养物质和病原体形成相同的感应系统,营养物质的摄入与病原体的入侵一样,除可引起代谢系统的反应外,还可像病原体一样诱发免疫系统的紊乱[17]。它涉及与经典炎症类似的分子和信号转导通路,也可造成多种炎性分子的表达和活性增强,而且这种炎症可以持续长期存在,在造成相关器官形态和功能损伤的基础上对机体的生理功能产生严重的影响[18]。Lee等[19]利用基因芯片检测了印度肥胖和非肥胖患者腹部脂肪组织细胞基因表达差异情况,结果表明,肥胖患者腹部脂肪细胞中大量与炎症和免疫相关的基因表达发生变化,其中52个表达上调,2个表达下调。这些基因中包括如肿瘤坏死因子α(TNF-α)、肿瘤坏死因子相关蛋白(C1QTNF5)、白介素1(IL-1B)和巨噬细胞移动抑制因子(MIF)等。Xie等[20]用高脂饲喂Wistar大鼠16周后诱发非酒精性脂肪性肝病(NAFLD),用基因芯片检测到了130多个基因上调,而涉及炎症的PPAR通路下调。在本研究发现实验组的8个通路中15个与免疫和炎症相关的基因表达均显著下调。非酒精性脂肪性肝病(NAFLD)发病机制理论“二次打击学说”[21]认为,一次打击诱发脂肪变性,随后在应激产生的细胞因子、持续存在的原有致病因素、肝星状细胞活化等作用下发生二次打击,导致肝脏发生炎症、坏死、细胞凋亡以及纤维化等。据此推测饮食诱导的高脂血症长爪沙鼠模型在造模型四周中存在两种可能,一种是处在一次打击期内,主要通过促使外周脂肪分解增加和胰岛素血症引起肝细胞脂肪堆积,所以机体表现只有高脂血症,肝脏脂肪变性,没有发生炎症[22],还有一种可能是发生了轻微的炎症,由于机体组织适应性反应机制的抗氧化、抗细胞调亡、瘦素的抗脂肪毒性等防御功能可与上述因素相抗衡继而机体自行修复[23],其过程大致是,首先与炎症相关的受体和信号通路增强如ECM受体的相互作用和趋化因子信号转导通路、细胞间粘附因子增多如细胞粘附分子、细胞因子与细胞因子受体相互作用、粘着等下调,然后炎症发生过程的相关代谢如白细胞跨内皮迁移、抗原处理和演示、补体和凝血级联也下调,表现为实验组长爪沙鼠肝脏因高脂血症发生炎症反应,继而发生抑制炎症与组织修复[24]。
本研究获得的上述差异基因与关键通路的功能需要继续进行相关验证,具体可以通过构建基因敲除或沉默载体来创建基因缺陷型细胞系(或动物模型),既可以进行单靶点操作,也可以多靶点操作,还可以利用转录组海量数据筛选出可用的微卫星标记,鉴定有功能的SNP,进而发展成为培育近交系的遗传标记。另外,由于在真核生物中,选择性剪切现象普遍存在,选择性剪接方式不同产生的功能基因不同,基因转录形成的mRNA 前体(pre-mRNA)过程中可形成不同的剪切异构体和基因表达,可能会对高脂血症的表型影响不同,因此寻找新的转录本和剪接方式也成为今后的研究主要方向之一。
参考文献:
[1] Wang J,Xian X,Huang W.Expression of LPL in endothelial-intact artery results in lipid deposition and vascular cell adhesion molecule-1 upregulation in both LPL and ApoE-deficient mice [J].Arterioseler Thromb Vasc Biol, 2007, 27:197-203.
[2] 赵冬. 中国人群血脂流行病学研究 [J].临床荟萃, 2006, 21(8):533-538.
[3] Kris-Etherton PM, Dietschy J. Design criteria for studies examining individual fatty acid effects on cardiovascular disease risk factors: human and animal studies [J]. Am J Clin Nutr, 1997, 65(suppl): 1590S-1596S.
[4] 赵严, 胡国勇, 柯爱武, 等. 高脂血症动物模型的研究进展 [J]. 中华胰腺病杂志, 2011, 10(4):25-27.
[5] Hegsted DM, Gailagher A. Dietary fat and cholesterol and serum cholesterol in the gerbil [J]. J Lipid Res, 1967, 8(3):210-214.
[6] 钟民涛, 王迎, 卢静, 等. 长爪沙鼠的高脂血症与动脉粥样硬化相关性分析 [J]. 中国比较医学杂志,2006, 16(6) :321-324.
[7] 徐黎明, 石巧娟, 徐磊, 等. 蒙古沙鼠非酒精性脂肪性肝病模型的建立与评价 [J]. 浙江预防医学, 2007, 19(11):1-2,7.
[8] Conesa A, Gotz S, Garcia-Gomez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research [J]. Bioinformatics, 2005, 21(18):3674-3676.
[9] Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium [J]. Nat Genet. 2000, 25(1):25-29.
[10] Ye J, Fang L, Zheng H, et al. WEGO: a web tool for plotting GO annotations [J]. Nucl Acids Res, 2006, 34:293-297.
[11] Kanehisa M, Goto S, Furumichi M, et al. KEGG for representation and analysis of molecular networks involving diseases and drugs [J]. Nucle Acids Res, 2010, 38:355-360.
[12] Wang L, Feng Z, Wang X, et al. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics, 2010, 26:136-138.
[13] http://www.kegg.jp
[14] Marioni JC, Mason CE, Mane SM, et al.RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays [J]. Genome Res, 2008, 18:1509-1517.
[15] Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq [J]. Nat Methods, 2008, 5:621-628.
[16] Pan Q, Shai O, Lee LJ, et al.Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing [J]. Nat Genet, 2008, 40:1413-1415.
[17] Demacker PN. The metabolic syndrome: definition, pathogenesis and therapy [J]. Eur J Clin Invest, 2007, 37(2): 85-89.
[18] Hotamisligil GS. Inflammation and metabolic disorders [J]. Nature, 2006, 444(7121): 860-867.
[19] Lee YH. Nair S, Rousseau E, et al. Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs non-obese Pima Indians: increased expression of inflammation-related genes [J]. Diabetologia, 2005, 48:1776-1783.
[20] Xie ZQ, Li HK, Wang K, et al. Analysis of transcriptome and metabolome profiles alterations in fatty liver induced by high-fat diet in rat [J]. Metabol Clin Exp. 2010, 59:554-560.
[21] Day CP, James OF. Steatohepatitis: a tale of two“hits”[J]. Gastroenterology, 1998, 114(4):842-845.
[22] 李巍, 石巧娟, 郭红刚, 等. 动态分析沙鼠非酒精性脂肪肝病形成及生化影响 [J]. 中国比较医学杂志,2011, 21(8):44-47, 52.
[23] 楼琦, 石巧娟, 郭红刚, 等. 非酒精性脂肪肝大鼠脂质代谢及病理变化的动态观察 [J]. 中国比较医学杂志,2012, 22(3):5-11.
[24] Wu G, Pfeiffer S, Scroder C, et al. Coagulation cascade activation triggers early failure of pig hearts expressing human complement regulatory genes [J]. Xenotransplantation. 2007, 14(1):34-47.
