染色质相互作用研究进展
2014-08-13潘有福
潘有福
(新加坡国立癌症中心转化研究实验室,新加坡 169612)
真核生物的染色体(质)在细胞核内,存在着不同的构象状态。当经历细胞分裂周期时,染色体呈现周期性的凝缩和去凝缩的过程。在间期的细胞核中,染色体也需要经过盘绕折叠,形成一种复杂的动态的高级结构。染色体一般以核小体作为基本的结构单位。在人的细胞核中,核小体是由147个DNA碱基对,缠绕在盘状的碱性组蛋白8聚体(H2A,H2B,H3,H4各2个)外围而形成。核小体之间还有一段连接DNA, 与H1组蛋白结合。每一条染色质线都是由众多核小体组成。这种念珠状的结构,经过多重折叠,并依附于染色体骨架上,形成更紧密高级的结构[1-2]。这种复杂的组织结构解决了将大约2 m长的DNA-组蛋白复合物压缩进直径约6μm的微小细胞核的组织结构问题,但同时也对如何将以线性方式携带的遗传信息在一定的时间和空间进行恰当表达提出了难题。因此了解染色体在细胞核内的组织结构及拓扑变化规律,以及这种结构变化对基因表达的调节控制,仍然是研究的热点之一。
近年发展起来的各种研究染色质相互作用的技术,尤其是染色体构象捕获技术(Chromosome conformation capture,3C),为我们研究染色体在细胞核中的三维构象提供了有力的技术手段,利用3C及其衍生技术,我们可以获得染色质上不同部位,以及染色质纤维之间在特定细胞状态下的相互作用信息。通过分析这些信息,我们可以了解间期细胞核中的染色体分布状态和特点,以及增强子和启动子相互作用以调控基因表达的规律。本文介绍用于研究染色质相互作用的技术,包括显微观察和3C及其衍生技术,回顾近年来利用相关技术取得的主要研究进展,并对该技术的应用前景作一讨论。
1 显微镜观察
染色体构象的研究技术主要包括传统的显微镜观察和近些年兴起的3C等技术(见表1)。显微镜观察主要是指荧光原位杂交(fuorescence in situ hybridization,FISH)技术。FISH技术通常利用荧光标记的DNA或RNA探针,来检测染色体上是否存在相应序列,或者细胞中是否存在特异的RNA分子。在染色质相互作用的研究中,如果同时利用两个或几个不同荧光标记的探针,就可以对染色质上两个或以上特异位点在细胞核内的分布进行定位,从而探讨不同位点的接近程度,获知有关染色体构象的信息。这一技术可以对待测位点的分布进行直观的显微观察,研究单个细胞中相关位点的空间位置关系[3-5]。
最近介绍的一个方法ChrISP(chromatin in situ proximity),是结合了FISH和原位邻位连接分析。这种临位连接分析,最初被用于检测两个蛋白质是否位于同一个复合体中(蛋白质相互作用)[6]。Chen et al[7]修饰了该方法,细胞先经甲醛固定,再与以地高辛或生物素标记的探针杂交,然后利用不同的一抗和二抗检测,其中二抗交联有不同的寡核苷酸序列。添加的长接头和荧光标记的短接头寡核苷酸,当两个位点间距离小于16.2 nm时,可形成一个荧光标记的环状DNA分子,从而得到阳性信号。类似的,电镜技术也可以用于观察单个细胞中两个已知位点的相互作用[8], 虽然分辨率很高,但是由于实验程序比较繁琐,所以较少应用。
由于受限于已知位点,单次实验中较少可用的探针,以及分辨率较低,尤其是仅限于研究已知位点的不足,FISH及相关技术一般难于用于检测未知位点的相互作用以及大规模的基因组水平的研究。但是FISH及其相关技术可为3C数据提供直观的佐证。
表1研究染色质相互作用的主要技术

技术简称原名检测位点检测手段主要文献FISH(荧光原位杂交)Fluorescence in situ hybridiza-tion两个或少数几个位点间荧光显微镜/共聚焦显微镜[3,5,7]3CChromosome conformation cap-ture两个或少数几个位点间PCR,琼脂糖电泳;定量PCR[9,10]4C“circular3C”or “chromosome conformation capture-onchip”特定位点与所有其他位点基因芯片[11,12]5CChromosome Conformation Capture Carbon Copy (5C)多个位点与多个位点基因芯片或下代测序技术[13]Hi-Ca high-throughput extension of 3C所有位点之间下代测序技术[14]ChIP-3C (ChIP-loop)Chromatin immunoprecipitati-on-3C/loop两个或少数几个位点间PCR,琼脂糖电泳;定量PCR[15,16]ChIA-PETChromatin Interaction Analy-sis by paired-end Tag se-quencing一个蛋白质介导的所有位点之间下代测序技术[5,17,18]
2 3C及其衍生技术
染色体构象捕获技术(3C)最初是从研究酵母染色体构象中建立起来的[8],后来在高等哺乳动物细胞株中得到应用和发展,如用于研究人基因组的一些基因座 beta球蛋白,IGF2 印记位点等[19-20]。这一系列技术的发展,是基于一个并不复杂的分子生物学技术,将物理距离靠近的DNA分子在原位进行限制性酶切和临位连接,再检测是否有异常DNA分子(线性距离较远或位于不同染色体上)形成。具体过程如下:对一个细胞群体进行甲醛固定,细胞经限制性内切酶消化或细胞悬浮液经超声波处理,得到平均长度为2~3个核小体长度的染色质丝。由于转录因子、转录辅助因子、RNA转录复合体等蛋白质之间以及与DNA特定序列之间存在着相互作用,因而形成蛋白质-DNA复合体。而这种相互作用的蛋白质-蛋白质-DNA复合体在适当条件下会在原位被固定下来。再对染色质片段进行连接,从而获得一个含有各种连接产物DNA片段的文库(3C文库),然后对重新形成的DNA片段进行鉴定、基因组比对和定量分析,获得染色质不同位点相互接近的频率,从而推断染色质的空间位置信息,绘制染色质相互作用图谱。
3C技术结合其他技术,已经发展出了4C、5C、CHIP-3C/Loop、ChIA-PET和Hi-C等技术(见表1),这些技术各有特点,后期所用的实验手段或处理程序不同,能够鉴定到的相互作用也不同。
常规3C技术可以用来检测两个或或少数几个位点间的相互作用(见表1)。细胞经固定,限制性内切酶或超声波处理,DNA片段经邻位连接(构象“捕获”),经去交联和DNA纯化,得到包含DNA片段两端自体连接或不同DNA片段异体连接的3C文库。3C文库DNA的检测主要通过设计位点特异的PCR引物,经PCR扩增后用琼脂糖凝胶电泳来检测,或者辅之以Sanger测序来确定,也可以用实时PCR来定量[9-10]。当DNA上线性距离较远的两个DNA片段,由于染色体空间构象变化而相互靠近,并被邻位连接而扩增得到阳性信号。这一方法结合FISH技术,可以为目标位点间的位置关系提供染色质相互作用的确切证据。其不足之处如上所述,由于依赖于特异性扩增(根据已知DNA序列设计引物进而对新形成的融合DNA片段扩增),所以只能研究少数已知位点间的染色质相互作用。
4C技术与常规3C基本相同,但部分克服了3C检测少数已知位点的不足。4C技术的一般策略是,细胞经固定后,首先利用限制性内切酶(识别4碱基或6碱基的内切酶)酶切,再用DNA连接酶进行邻位连接,用于检测特定的一个已知位点(诱饵位点)与其他位置位点间的相互作用。由于在4C文库的构建过程中会有一些环状DNA形成(与3C类似),因而可以利用已知位点设计引物,通过反向PCR扩增得到与该诱饵位点相互作用的其他染色质位点信息。4C文库可以通过下代测序技术或基因芯片技术来检测[21]。4C技术可以比常规3C技术得到更多的染色质相互作用信息,适合于对某一基因座的未知调控元件进行鉴定或对该染色质位点所参与的空间构象进行探讨(见表1)。
5C技术因为类似于复制了部分3C文库(3C-Carbon Copy),因而称为5C,是基于3C和连接介导的扩增(ligation-mediated amplification,LMA)技术。5C技术是在构建3C文库的基础上,通过精心设计两套引物序列,利用Tag连接酶连接两个与文库中DNA精确杂交的引物序列,再通过通用引物序列的扩增,获得3C文库中部分相互作用的染色质位点信息,详细步骤请见[13]。5C曾用于研究beta球蛋白基因座与调控序列间的染色质相互作用,由于需要设计两套引物,并受限于研究特定的DNA区域,所以应用比较有限。
如果说3C、4C和5C 技术都是探讨已知位点与已知或未知的位点的相互作用,从而探讨染色体的局部构象,那么在此基础上建立的Hi-C技术则可以用来检测基因组水平的部分或所有未知位点之间的相互作用。Hi-C方法也是在3C技术的基础上建立的。细胞经固定和限制性内切酶酶切,形成的5’突出末端用dNTP(包含生物素化的dCTP)填充,在稀释的情况下将平头末端再连接。产物经超声波处理,含生物素的DNA片段经亲和素沉淀(见图1)。制得的Hi-C文库,再经双端高通量测序,分析和基因组序列比对,检测全基因组染色质间的相互作用位点[14]。Hi-C方法是在基因组水平研究所有染色质位点间可能存在的染色质相互作用关系的技术。获得的信息非常丰富,一般信号噪音比较高。经过处理后的信息,可以和5C数据一样以二维热点图(heat map)的方式来呈现[14]。

细胞经固定,限制性内切酶酶切,末端用dNTP(包含生物素化的dCTP)填充,连接,超声波处理,亲和素沉淀,双端高通量测序,分析和基因组序列比对。红线和蓝线表示DNA分子,各种颜色的椭圆表示与DNA结合的各种蛋白质,如深绿色椭圆表示雌激素受体,较大的黄色椭圆表示RNA聚合酶II,黑色线段表示接头或限制性酶切位点(连接后),其上的绿色小圆表示生物素分子。图示两个发生高频率染色质相互作用的区域。图1 Hi-C的主要流程
ChIP-3C或ChIP-loop技术则是结合了ChIP和3C技术,可以研究某一蛋白质介导的染色质之间相互作用,如雌激素受体或RNA聚合酶II等转录因子介导的增强子-启动子间的相互作用[5,16]。ChIP-3C一般是在利用某一特异性抗体进行染色质免疫沉淀的过程中,对免疫复合物中的DNA片段进行邻位连接,再经去交联和DNA纯化,可以得到特定蛋白质介导的染色质相互作用的3C文库。其检测方法与前述3C文库的相同。不足之处是对于每一个染色质间的相互作用,都需要设计相应的引物来扩增并分析。
同Hi-C技术一样,ChIA-PET技术也是在基因组水平检测全新的染色质相互作用的技术。ChIA-PET是我们首先用雌激素受体阳性的人乳腺癌细胞MCF7建立起来的,后来在小鼠、干细胞等材料中也得到了应用[5,17]。利用不同的ChIP 级的特异性抗体,针对雌激素受体和RNA聚合酶II等,可以探测到以该被检测蛋白为锚定点介导的基因组水平染色质相互作用。ChIA-PET技术与ChIP-3C技术在前期的样品制备过程基本相同,如细胞经固定、超声波处理和染色质免疫沉淀。此后,ChIP产物在稀释后(目的是为了降低非特异性临位连接产物),连接生物素化的接头(含MmeI位点),DNA-蛋白质复合体经65℃去交联,DNA经纯化,MmeI消化,亲和素交联的磁珠沉淀。制得的ChIA-PET文库,经高通量测序,分析和基因组序列比对,就可以绘制如雌激素受体介导的全基因组染色质纤维相互作用的位点(见图2)。ChIA-PET通过鉴定增强子-启动子间相互作用而形成的环状结构,可以同时鉴定某一时刻所有与目标基因位置靠近的增强子或其他DNA调控元件[5,17]。

细胞经固定,超声波处理,抗体免疫沉淀,用生物素化的ChIA-PET接头(含MmeI位点)连接。再经MMeI酶切,用链霉亲合素交联的珠子纯化重新连接的DNA分子。然后加上双末端测序接头,扩增,测序,基因组比对,鉴定蛋白质在DNA上的结合位点及染色质长程相互作用。红线和蓝线表示DNA分子,各种颜色的椭圆表示与DNA结合的各种蛋白质,如深绿色椭圆表示雌激素受体,较大的黄色椭圆表示RNA聚合酶II,黑色线段表示接头,其上的绿色小圆表示生物素分子。图示检测到的两个染色质位点间的相互作用。图2 ChIA-PET的主要流程
ChIA-PET与Hi-C的主要不同点有:一是Hi-C没有ChIP这一步,因而不受限于ChIP级别的特异性抗体;二是两者在初次连接DNA片段时所用的接头不同,因而制备文库的策略不同;三是ChIA-PET只能检测到细胞群体中以与DNA结合的某一蛋白质所介导的所有的染色质位点间的相互作用,而Hi-C理论上可以捕捉到一定的细胞群体中所有的染色质位点间的相互作用,包括转录因子介导的以及染色质与染色质间直接的相互作用。对于染色质上不同位点间的相互作用频率而言,由于同一染色质上的顺式相互作用远远高于不同染色质上的反式相互作用(cis-trans ratio ,CTR),以及作用强度随距离而衰减(distance-dependent decay, DDD),这就使得Hi-C技术的分辨率目前只能达到0.1到1 Mb[14]或者5-10kb[22]。而ChIA-PET通过染色质免疫沉淀一步消除了大量非特异性的相互作用信号,所以理论上的分辨率应该为实验中能区分的DNA上临近的两个蛋白质结合位点。3C技术结合目前的高通量下代测序技术,已经逐渐成为研究染色质间相互作用的主流方向。因为ChIA-PET和Hi-C技术是目前在基因组水平探讨和鉴定染色质长程相互作用的最主要技术,因此我们重点回顾利用这两个技术获得的主要进展。
3 染色体构象与基因表达及染色体三维构象
3.1 雌激素受体介导的染色质间相互作用 雌激素受体(EstrogenReceptors,ERs)是核受体家族的一员,ERs在发育、正常生理、病理过程中起着重要作用,因此研究ERs介导的基因调控机理对于深入研究相关疾病具有重要意义。最初不同研究组利用不同的方法对雌激素受体α(ERα)在基因组中的结合位点进行定位时,发现大多数结合位点都分布在远离基因启动子的5kb以上的地方[23-24],因此如何界定这些潜在的调控元件所控制的特定基因,成为研究雌激素信号通路的一个挑战。以TFF基因为例,我们证实在其上游10.5kb的一个ERα结合位点在促进TFF1基因的表达中起着关键作用,通过形成增强子-启动子环状结构相互作用,从而促进其高表达[16,25]。常规的这类研究DNA调控序列的方法,每次只能针对少量位点,比较费时费力。为了探讨这些基因组范围内ERα结合位点所调控的基因,CHIA-PET技术应运而生[5]。利用MCF7 细胞,在初次实验中从两个文库中各鉴定到1475个和3561个相互作用,前100个相互作用中有80% 是相同的。总共鉴定了至少689 ERα 结合的染色质相互作用区域(两次实验能重复得到的数据。这一方面表明染色质相互作用的动态性,一方面表示那些强的相互作用相对而言比较容易被捕捉到。随着进行更深度的测序,已检测到更多的相互作用(未发表数据)。对于鉴定与基因转录调控有关的未知的DNA调控元件来说,CHIA-PET技术目前而言仍是最为高效的方法。ERα介导的染色质相互作用与基因表达有着密切的关系。在锚定点附近的基因,多数呈活跃表达状态,而环内基因则呈现被抑制趋势。所以这种染色质相互作用,可以促进一些基因的表达,同时也抑制另外一些基因的活性[5,26]。
另一发现是,ERα与特异的雌激素反应元件的结合及其介导的染色质相互作用,需要FoxA1和AP2γ的参与[27-28]。61%的雌激素受体结合位点有FoXA1和AP2γ[28]。在这种雌激素介导的信号传导过程中,AP2γ可能和FoxA1一样,起着前驱因子的作用。所以目前我们认为ER介导的染色质长程相互作用的机理,可能如图3所示[28]。
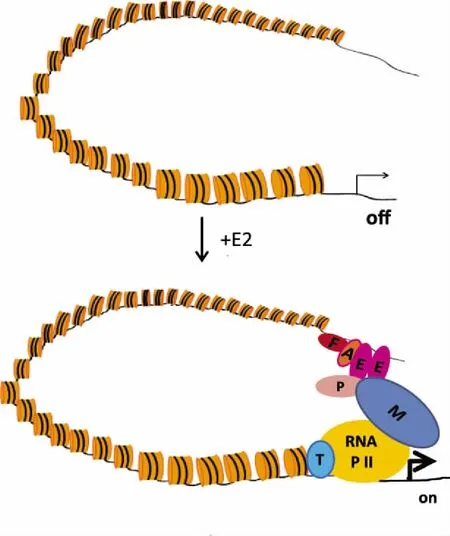
没有雌激素(E2)时,ERα调控的基因不表达或低表达。当有E2存在时,E2进入细胞质,与ERα结合,进入细胞核内,与FoxA1(F)和AP2γ(A)等作用结合在雌激素反应元件上,并与其他转录因子作用,与启动子发生染色质相互作用,激活RNA聚合酶II(RNAP II)的转录活性,从而促使目标基因表达。P,p300/CBP;T,TFIID;M,Mediator。 图3 雌激素受体ERα介导的染色质长程相互作用模型
3.2 RNA聚合酶II介导的染色质间相互作用 RNA聚合酶II对mRNA,snRNA和许多microRNA的转录起着关键作用。各种转录因子如ERα等介导的染色质长程相互作用,很多都和启动子区的RNA聚合酶II有关,所以基因组水平的RNA聚合酶II介导的染色质长程相互作用也是很重要的一方面,这种相互作用最初在MCF7中得到了确认。起初鉴定到了RNA 聚合酶II介导的12,626相互作用,随着深度测序,得到了成倍的——24,126个相互作用[18]。Li 等探讨了包括MCF7等5种细胞株中RNA 聚合酶II相关的启动子可以作为染色质相互作用的关键锚点。而启动子-启动子相互作用对于多数临近基因的表达有促进作用[18]。这表明传统意义上的启动子在染色质相互作用的环境下可以在某种程度上扮演增强子的作用。
类似的发现也存在于3种小鼠细胞类型中,并且与干细胞的分化过程有关。Zhang 等[29]比较了处于不同分化程度的3种小鼠细胞:胚胎干细胞(embryonic stem cells ,ESCs), 神经干细胞(neural stem cells,NSCs)和神经祖细胞(neurosphere stem/progenitorcells,NPCs)中RNA聚合酶II介导的相互作用,总共检测到40 000 个染色质长程相互作用。当启动子定义为距离转录起始位点+/- 2.5碱基对时,一半的染色质相互作用表现为启动子-启动子之间,另外的表现为启动子-基因内增强子或启动子-基因间增强子。这种相互作用表现出很强的细胞特异性。并在3种细胞株中分别鉴定了8,309, 4,463 和3,649个潜在的远距离DNA调控元件。
Kieffer-Kwon等也有类似的发现,小鼠多能胚胎干细胞和已分化的B淋巴细胞基因组中有不同的增强子和启动子之间相互作用的图谱,而且也发现胚胎干细胞和已分化的B淋巴细胞中,活跃表达的基因,例如Myc和Pim1等,在两种细胞中使用着不同的增强子组。这也表明增强子和启动子的相互作用,增加了基因转录调控的复杂性并表现出细胞发育阶段的特异性[30]。但是对于外源激素刺激的信号传导过程研究,表明这种增强子-启动子之间的染色质位点间的相互作用,在外源激素刺激前,就已经存在。Jin等用Hi-C研究了TNF-α 信号通路中,NF-κB介导的启动子-增强子的相互作用。他们发现这种相互作用的成环结构,在TNF-α 信号传递前就已存在。而且这种相互作用也在IFN-γ, TNF-α, β-estradiol和5α-dihydrotestosterone处理的不同细胞中预先存在[22]。这与我们早期在雌激素(β-estradiol)介导的染色质相互作用中的研究并不完全一致。造成这种差异的原因以及这种预先成环结构的存在是否具有普遍意义,值得进一步探讨。
3.3 CTCF介导的染色质间相互作用 早期的研究发现,CTCF(CCCTC-binding factor,CTCF)是特异性地与DNA上的CCCTC序列结合的蛋白,因为可以阻断增强子对基因的作用,具有绝缘子或隔离子特性,所以又称为转录阻抑蛋白(Transcriptional repressor CTCF)。CTCF在基因转录调控,染色质三维结构的组织中起着十分关键的作用[31-32]。Handoko等用ChIA-PET技术,研究了CTCF在小鼠胚胎多能干细胞中的结合位点和介导的染色质相互作用[31]。结果表明CTCF在建立和维持染色质的局部构象,调节基因簇的协同表达,介导DNA调控元件与启动子的相互作用,以及染色体局部的构象等方面发挥着重要作用。他们综合所有CTCF的ChIA-PET文库,得到了1480个顺式和336个反式染色质相互作用。一部分CTCF介导的染色质相互作用锚定点与p300的结合位点一致,而p300是转录激活因子。降低CTCF蛋白质水平及其介导的染色质相互作用,相应降低了该区域的基因转录水平,从而揭示在一些情况下CTCF可以阻断增强子的活性,而有时也可以通过介导这种相互作用,使p300等转录因子与特定基因的启动子发生长程相互作用,从而激活基因的转录活性。DNA的核纤层结合域(Lamin-associated domains,LADs)两侧,常见CTCF环状结构。CTCF通过扮演隔离子的角色,限定LADs局部区域,使LAD区域内的基因呈现低表达状态。同时,CTCF的分布与组蛋白的修饰也密切相关,如H3K4me1和H3K27me3的分布与 CTCF的结合位点高度相关,H3K36me3 呈高度负相关[33-34]。类似的结果也在SMC1A,一个Cohesin复合体的亚基参与的小鼠研究中被证实[35]。Cohesin与CTCF常相互作用,是功能密切相关的蛋白质。Cohesin介导的相互作用影响细胞干性基因表达,与维持细胞的多能性密切相关[36-37]。因而Cohesin/CTCF介导的染色质相互作用是在不同组织中基因特异表达的一种基本调控机制。
Zuin et al[38]2013 探讨了CTCF和Cohesin复合体在染色体高级组织结构中的作用,证实与CTCF 和Cohesin对于染色体拓扑结构有不同的作用。Cohesin更多参与距离较短的染色质位点间的相互作用,而CTCF既参与距离较短的染色质位点间的相互作用,也参与临近拓扑结构域之间的相互作用。分别降低两者的蛋白质水平会引起不同的染色体拓扑结构的变化,也相应引起了不同的基因表达谱的变化。
如果比较CTCF和ERα介导的染色质相互作用,我们会发现二者介导的染色质相互作用对基因的转录调控,并非单一方向的,而是具有双向调节作用。既可以抑制被调控的基因,也可以促进一些基因的活性。另外ERα介导的染色质长程相互作用形成的环内基因有一定程度的抑制,也说明雌激素受体除了作为积极参与转录调控以外,还可以通过介导染色质的相互作用而表现出和CTCF相同的功能,即对染色体进行区域化分隔,从而在染色体高级结构层次对基因表达进行调控。
3.4 染色质相互作用与染色质在细胞核内的空间组织 关于染色质丝在细胞核内的组织,有多种模型被提出,如有分形小球(fractal globule),平衡小球(equilibrium globule)等模型,其中染色质丝的分形小球模型获得了一些实验数据的支持[39]。分形小球模型主要指多聚体在凝聚而形成的一种致密的状态,由于拓扑结构的限制,不会在链内部形成“结状”结构。Dekker研究组根据以上这些相互作用的频率及其位点间的距离判断,在百兆碱基对的尺度上,染色质线在细胞核中的分布更符合分形球状模型,而不是以前认为的平衡球状模型[14]。
关于染色体在细胞核内的组织以及对于染色体不同结构域的定义仍没有统一。但一般认为,每条染色体在细胞核内占据着一定的区域,而每条染色体由于不同的结构域组成。染色体不是随机杂乱分布的,而是遵循着一定的规律[40-43]。如Cremer 等认为,在间期细胞核中,染色体领域(chromosome territories, CTs)是一个细胞核结构的一个基本的特征[40]。对少量的染色体部分的研究表明,尺度大约为1Mb的染色体片段是组成染色体域的基本单位[40,44]。他们认为染色体领域在光镜下表现为吉姆萨染色后的深染染色质团块。而这些1Mb的染色质域又是由小的环状染色质域组成的[40]。
而最近Dekker和Lander 等合作用lymphoblastoid和K562细胞得到了染色体长程相互作用资料[42]。他们认为每条染色体在细胞核内占据着一定的区域(Chromosome Territories,CTs),即每条染色体占据着的核内区域为其染色体领域(图4 扇形部分)。同时,每条染色体从空间分布上可以大致分为两种次级结构域——区隔(compartment A or B)。其中一种区隔基因密度较高,染色体多呈开放状态(见图4中A区隔);而另一类基因密度较低,染色体构象较致密(见图4中B区隔)。有数据表明大约5Mb的区隔A和B沿着染色质丝分布,区隔间可以发生染色质的相互作用,一般A倾向于与A作用,B倾向于与B作用,虽然来自不同区隔中的位点相互作用频率较低。而区隔是由若干个染色体拓扑联合域(topologically associating domains ,TADs)组成。每个区隔内的一系列染色质位点可以发生频率较高的相互作用(见图4)。

图示间期细胞核中一条染色体的局部放大。扇形区域表示一个放大的染色体领域(CT)。圆A和B表示染色体的两个相邻区隔(CC),其中A表示比较活跃的基因比较多的区域,而B表示不太活跃基因密度较低的部分,分别大概5Mb左右。A和B分别有数个拓扑联合域(TADs)组成,每个拓扑联合域约500kb。其中的部分拓扑结构域用红色或蓝色圈表示(CTCF在全基因组都有分布,但在拓扑联合域之间有较多分布,图中未标示)。图4 间期细胞核中染色质的局部构象
21号染色体大小为48Mb,如果假设每个拓扑联合域为500kb,每10个拓扑联合域组成一个区隔,则最小的21号染色体就由近10个区隔组成。而最大的1号染色体(249Mb)由大约50个区隔组成。当然实际情形要比这复杂得多,不仅因为这些拓扑联合域和区隔等次级域的大小不是均匀的,而且因为染色体也是动态的。
4 3C及其衍生技术的应用前景
3C技术自从建立以来,已经发展得比较成熟了,尤其以ChIA-PET和Hi-C技术为代表,但也有一些不足之处。由于这些结果都是来自于许多细胞组成的样品,即基于对细胞群体的实验和统计分析,因而难于估计不同细胞中的即时的染色质相互作用和染色质组织特点。如何解析单个细胞中的染色质相互作用和染色体的空间组织,以及在发育过程中,或者疾病发生发展过程中染色质相互作用和基因组结构的动态变化,仍是研究的难点。另外,3C技术由于临近染色体的随机靠近,捕获的信号会湮没那些特征性的比较稳定的染色质位点间的联系。尤其是如何降低Hi-C技术的噪音,提高灵敏度也是需要关注的。不过,尽管有一些不足,3C技术仍然是很有用的基因组学研究工具。
首先,可以通过特定的转录相关蛋白质的免疫沉淀,用于鉴定未知的与基因调控表达有关的重要序列,是否与基因启动子区存在着相互作用。当然,利用3C技术获得的染色质相互作用,对于他们是否具有增强子的作用或其他功能,还需要通过其他实验,如荧光素酶报告基因实验,转基因斑马鱼、TALENs或CRISPR/Cas等基因组编辑工具来检验。
其次,全基因组水平的染色质特异以及非特异的相互作用数据,将会为进一步解析染色质在细胞核内的组织结构提供丰富的资料。如肿瘤的发生发展一般是一个长期复杂的过程,不仅与若干个基因突变有关,在某些肿瘤中,还常伴随着染色体结构的改变,如染色体倍性的改变,染色体片段的增加,缺失及易位,基因的扩增等。这些染色体一级结构的改变,是否以及如何影响到染色体高级构象的变化,是一个值得探讨的问题。如在ICF综合症(Immunodeficiency-centromeric instability-facial anomalies syndrome)中, DNA甲基化的改变影响到了染色体领域的变化。类似的, 3C技术无疑也可以用于研究在一些病理变化过程中,染色体的三维构象是否呈现特异性的的变化,是否可以成为相关疾病的特征性标志。
另外,3C数据不仅可以用于研究染色体的高级结构,也可以用于研究基因组的线性序列。如最近Kaplan等[45]利用染色质相互作用强度表现为“顺式”和“近距离较强”的特性,利用Hi-C技术预测和拼接一些尚未连接的人类基因组或其他基因组的缺口区域。
最后,这些技术也可以用于单核苷酸多态性(single nucleotide polymorphisms ,SNPs)的相关研究。比如可以通过探讨有统计学意义的与疾病相关的已知SNPs位点,是否参与染色质位点之间的相互作用,进而参与基因的调控,从而鉴别基因本体以外的与疾病相关的SNPs并探讨其可能的功能。
总之,随着未来建立在3C基础之上的实验技术的继续发展和应用,以及相应计算技术的开发,我们无疑会得到丰富的染色质相互作用的资料,这些资料必将极大促进功能基因组的研究。
[参考文献]
[1] Luger K, Mader A W, Richmond R K, et al.Crystal structure of the nucleosome core particle at 2.8 A resolution [J]. Nature, 1997,389(6648):251-260.
[2] Olins D E, Olins A L. Chromatin history: our view from the bridge[J].Nat Rev Mol Cell Biol,2003,4(10):809-814.
[3] Lanzuolo C, Roure V, Dekker J, et al. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex[J]. Nat Cell Biol,2007, 9(10):1167-1174.
[4] Matarazzo M R, Boyle S, D’Esposito M,et al. Chromosome territory reorganization in a human disease with altered DNA methylation[J].Proc Natl Acad Sci USA,2007,104(42):16546-16551.
[5] Fullwood M J, Liu M H, Pan Y F, et al.An oestrogen receptor α-bound human chromatin interactome[J].Nature,2010, 462(7269):58-64.
[6] Söderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation[J].Nat Methods,2006,3(12):995-1000.
[7] Chen X, Shi C, Yammine S, et al. Chromatin in situ proximity (ChrISP): single-cell analysis of chromatin proximities at a high resolution[J].Biotechniques,2014,56(3):117-124.
[8] Su W, Porter S, Kustu S, et al. DNA-looping and enhancer activity?: Association between DNA-bound NtrC activator and RNA polymerase at the bacterial ginA promoter[J].Proc Nati Acad Sci USA,1990,87(14):5504-5508.
[9] Dekker J, Rippe K, Dekker M, et al. Capturing chromosome conformation[J].Science,2002,295(5558):1306-1311.
[10] Hagège H, Klous P, Braem C, et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR)[J]. Nat Protoc,2007, 2(7):1722-1733.
[11] Simonis M, Kooren J, de Laat W. An evaluation of 3C-based methods to capture DNA interactions[J].Nat Methods,2007, 4(11):895-901.
[12] Zhao Z, Tavoosidana G, Sjölinder M, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions[J].Nat Genet,2006, 38(11):1341-1347.
[13] Dostie J, Richmond T A, Arnaout R A, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements[J].Genome Res,2006, 16(10):1299-1309.
[14] Lieberman-Aiden E, van Berkum N L, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome[J].Science,2009, 326(5950):289-293.
[15] Horike S, Cai S, Miyano M, et al. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome[J].Nat. Genet,2005, 37(1):31-40.
[16] Pan Y F, Wansa K D, Liu M H, et al. Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements[J].J Biol Chem,2008,283(47):32977-32988.
[17] Li G, Fullwood M J, Xu H, et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing[J].Genome Biol,2010, 11(2):R22.
[18] Li G, Ruan X, Auerbach R K,et al.Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation[J].Cell,2012 ,148(1-2):84-98.
[19] Tolhuis B, Palstra R J, Splinter E, et al.Looping and interaction between hypersensitive sites in the active beta-globin locus[J]. Mol Cell,2002,10(6):1453-1465.
[20] Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops[J].Nat Genet,2004, 36(8): 889-893.
[21] Wei Z, Gao F, Kim S, et al. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency[J].Cell Stem Cell ,2013,13(1):36-47.
[22] Jin F,Li Y, Dixon J R, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells[J].Nature,2013 ,503(7475):290-294.
[23] Carroll J S, Meyer C A, Song J, et al. Genome-wide analysis of estrogen receptor binding sites[J].Nat Genet,2006, 38(11):1289-1297.
[24] Lin C Y, Vega V B, Thomsen J S, et al.Whole-Genome Cartography of Estrogen Receptor α Binding Sites[J].PLoS Genet,2007, 3(6): e87.
[25] Theodorou V, Carroll J S. Estrogen receptor action in three dimensions - looping the loop[J].Breast Cancer Res,2010, 12(1):303.
[26] Chepelev I, Wei G, Wangsa D, et al. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization[J].Cell Res,2012, 22(3):490-503.
[27] Carroll J S, Liu X S . Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1[J].Cell,2005, 122(1):33-43.
[28] Tan S K, Lin Z H, Chang C W, et al. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription[J].EMBO J,2011, 30(13):2569-2581.
[29] Zhang Y,Wong C H,Birnbaum R Y, et al.Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations [J].Nature,2013,504(7479),306-310.
[30] Kieffer-Kwon K R,Tang Z,Mathe E,et al.Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation[J]. Cell,2013, 155(7):1507-1520.
[31] Cuddapah S, Jothi R, Schones D E, et al.Global analysis of the insulator binding proteinCTCFin chromatin barrier regions reveals demarcation of active and repressive domains[J]. Genome Res,2009, 19(1):24-32.
[32] Ong C T, Corces V G.CTCF: an architectural protein bridging genome topology and function[J].Nat Rev Genet,2014, 15(4):234-246.
[33] Handoko L, Xu H, Li G, et al. CTCF-mediated functional chromatin interactome in pluripotent cells[J].Nat Genet,2011, 43(7):630-638.
[34] Lee B K, Iyer V R. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation[J]. J Biol Chem,2012, 287(37):30906-30913.
[35] De Mare L E, Leng J, Cotney J, et al. The genomic landscape of cohesin-associated chromatin interactions[J].Genome Res,2013, 23(8):1224-1234.
[36] Zhang H, Jiao W, Sun L, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming[J].Cell Stem Cell ,2013, 13(1):30-35.
[37] Sexton T, Cavalli G. The 3D genome shapes up for pluripotency[J].Cell Stem Cell .2013, 13(1):3-4.
[38] Zuin J, Dixon J R, van der Reijden M I, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells[J].Proc Natl Acad Sci USA,2014, 111(3):996-1001.
[39] Mirny L A. The fractal globule as a model of chromatin architecture in the cell[J].Chromosome Res,2011, 19(1):37-51.
[40] Cremer T, Cremer M. Chromosome territories[J].Cold Spring Harb Perspect Biol ,2010, 2(3):a003889.
[41] Marti-Renom M A, Mirny L A. Bridging the resolution gap in structural modeling of 3D genome organization[J]. PLoS Comput Biol,2011, 7(7):e1002125.
[42] Dekker J, Marti-Renom M A, Mirny L A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data[J].Nat Rev Genet,2013, 14(6):390-403.
[43] Gibcus J H, Dekker J. The hierarchy of the 3D genome[J].Mol Cell ,2013, 49(5):773-782.
[44] Cope N F, Fraser P, Eskiw C H. The yin and yang of chromatin spatial organization[J].Genome Biol,2010, 11(3):204.
[45] Kaplan N, Dekker J. Nat Biotechnol. High-throughput genome scaffolding from in vivo DNA interaction frequency[J].Nature Biotechnol,2013, 31(12):1143-1147.
