2-芳基-4-戊烯基酯的“一锅法”合成
2014-08-07柏一慧杜金艳
柏一慧, 杜金艳
(浙江师范大学 化学与生命科学学院,浙江 金华 321004)
2-芳基-4-戊烯基酯是重要的有机合成中间体之一[1],它具有芳基、烯丙基和酯基3种不同的官能团,可以通过简单的化学反应转变成其他有用的有机结构单元;而且,2-芳基-4-戊烯基酯含有一个手性中心,该手性原子可以进一步引入到具有生理活性的、手性的天然产物和药物分子中.因此,2-芳基-4-戊烯基酯的合成具有重要意义.[3,3]-重排反应是一种常见的制备2-芳基-4-戊烯基酯的方法[2],但是该反应往往需要剧烈的反应条件(例如很高的反应温度)才能进行,同时重排反应产率也不够理想.所以,进一步发展简单、高效的2-芳基-4-戊烯基酯的合成方法非常必要.
最近,Tsuji-Trost反应[3]在有机合成方面得到了广泛应用和发展,成为构建含有烯丙基的化合物的有效途径.显然,钯催化酯的烯丙基化反应是合成2-芳基-4-戊烯基酯的最直接、有效的方法之一.但是,这一策略存在2个问题:1)酯的α-位碳原子酸性较小,常常需要二异丁基胺基锂(LDA)等强碱才能脱质子;2)酯在强碱性条件下容易发生缩合等副反应,从而导致产率较低.正因为如此,钯催化酯的α-烯丙基化反应还很少见报道.对此,笔者的想法是将酯转变成更为稳定的酰基苯并三唑,避免原料的分解;另外,苯并三唑的引入可以提高羰基α-位碳原子的酸性,增加碳负离子亲核试剂的浓度,这样就有可能有利于钯催化的羧酸衍生物α-烯丙基化反应的进行.通过研究,本文实现了一个钯催化的α-芳基乙酰基苯并三唑烯丙基化/酯化串联反应,为2-芳基-4-戊烯基酯的合成提供了一种新途径.需要指出的是,苯并三唑[4]可以在温和的条件下被其他亲核试剂置换,最终为2-芳基-4-戊烯基酯的“一锅法”合成奠定了基础.
1 实验部分
1.1 试剂
苯乙酰氯、对氯苯乙酰氯、对甲氧基苯乙酰氯、苯并三唑、烯丙醇、氯甲酸乙酯、三乙胺、醋酸钯、双(二亚芐基丙酮)钯、三苯基膦和1,2-双(二苯基膦)乙烷等试剂皆为分析纯.
1.2 实验仪器
集热式磁力搅拌器、旋转蒸发仪(郑州长城科工贸有限公司);Bruker Advance 400 或600 MHz核磁共振仪.
1.3 实验方法
1.3.1α-芳基乙酰基苯并三唑的制备
α-芳基乙酰基苯并三唑以α-芳基乙酰氯和苯并三唑为初始原料,在二氯甲烷中室温反应过夜制得(见图1).

图1 α-芳基乙酰基苯并三唑的制备
在250 mL三颈烧瓶中加入苯并三唑(5.95 g,50 mmol)和干燥的二氯甲烷100 mL,缓慢加入含不同取代基的苯乙酰氯60 mmol,室温搅拌过夜.旋转蒸发除去溶剂后得到白色固体,然后用乙酸乙酯和石油醚重结晶,分别得到化合物1a,1b和1c.
化合物1a:产率85%,白色固体,熔点60~62 ℃.1H NMR(600 MHz,CDCl3)δ:8.29(d,J=8.2 Hz,1H),8.15(d,J=8.3 Hz,1H),7.68~7.64(m,1H),7.50(d,J=7.4 Hz,2H),7.40(t,J=7.7 Hz,2H),7.37~7.33(m,2H),4.76(s,2H);13C NMR(150 MHz,CDCl3)δ:170.2,146.2,132.4,131.1,130.4,129.8,128.7,127.6,126.2,120.0,114.4,41.9.
化合物1b:产率89%,白色固体,熔点92~94 ℃.1H NMR(600 MHz,CDCl3)δ:8.29(d,J=8.2 Hz,1H),8.15(d,J=8.3 Hz,1H),7.67(t,J=7.9 Hz,1H),7.53(t,J=7.7 Hz,1H),7.41(d,J=8.5 Hz,2H),6.93(d,J=8.5 Hz,2H),4.70(s,2H);13C NMR(150 MHz,CDCl3)δ:170.6,159.1,146.3,131.3,130.9,130.4,126.2,124.4,120.1,114.5,114.3,41.2.
化合物1c:产率88%,白色固体,熔点94~96 ℃.1H NMR(600 MHz,CDCl3)δ:8.28(d,J=8.2 Hz,1H),8.14(d,J=8.3 Hz,1H),7.65(t,J=8.0 Hz,1H),7.51(t,J=7.9 Hz,1H),7.41(d,J=8.7 Hz,2H),6.92(d,J=8.7 Hz,2H),4.69(s,2H),3.81(s,3H);13C NMR(150 MHz,CDCl3)δ:170.5,159.0,146.2,131.2,130.9,130.4,126.2,124.4,120.1,114.4,114.2,55.2,41.1.
1.3.2 烯丙基碳酸乙酯的合成
烯丙基碳酸乙酯以烯丙醇和氯甲酸乙酯为原料,在三乙胺的存在下室温反应过夜制得(见图2).
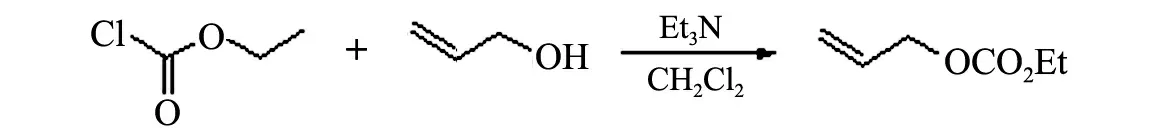
图2 烯丙基碳酸乙酯的制备
在250 mL三颈烧瓶中加入烯丙醇(5.6 g,100 mmol)、三乙胺(12.1 g,120 mmol)和150 mL干燥二氯甲烷,冰浴下加入氯甲酸乙酯(11.4 g,105 mmol),室温反应过夜.反应体系加100 mL水淬灭,二氯甲烷(50 mL)萃取,有机相用饱和碳酸氢钠溶液洗、无水硫酸钠干燥,常压蒸馏得烯丙基碳酸乙酯(8.9 g,产率82%)无色液体,沸点132~134 ℃.1H NMR(CDCl3,400 MHz)δ:1.32(t,J=7.2 Hz,3H),4.20(q,J=7.2 Hz,2H),4.55~4.60(m,2H),5.35~5.37(m,2H),5.86~6.02(m,1H).
1.3.3 2-芳基-4-戊烯基酯的“一锅法”合成
在25 mL反应管中,氮气气氛下,加入1,2-双(二苯基膦)乙烷(4.0 mg,0.01 mmol)、双(二亚芐基丙酮)钯(5.7 mg,0.01 mmol)和四氢呋喃2 mL,接着加入合成的烯丙基碳酸乙酯(39 mg,0.30 mmol)和α-芳基乙酰基苯并三唑(0.25 mmol).反应体系在50 ℃搅拌2 h后,加入1 mL MeOH继续搅拌2 h,接着加5 mL水淬灭,乙酸乙酯(5 mL)萃取3次,有机相合并后无水硫酸钠干燥,硅胶(300~400目)柱色谱分离纯化(洗脱液:V(石油醚)/V(乙酸乙酯)=15/1),得无色液体产物(见图3).
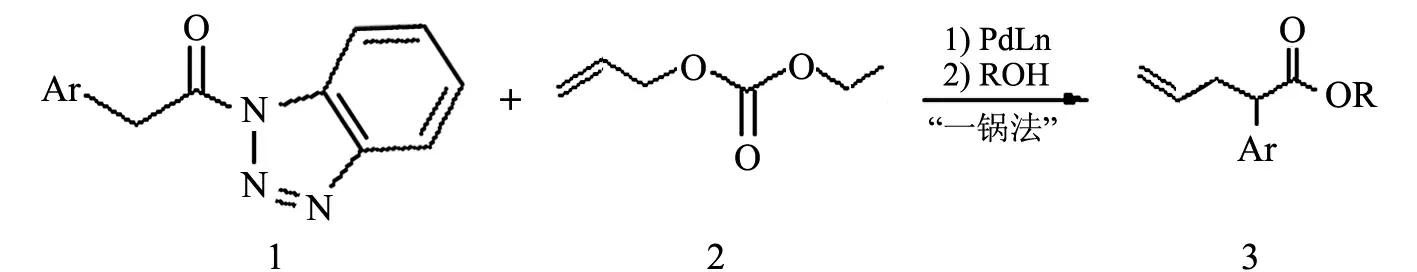
图3 2-芳基-4-戊烯基酯的“一锅法”合成
2 结果与讨论
2.1 钯催化的烯丙基化反应条件优化
笔者对α-苯乙酰基苯并三唑和烯丙基碳酸乙酯的Tsuji-Trost反应条件进行了研究.
首先,以双(二亚芐基丙酮)钯为催化剂对配体进行了筛选.结果发现:以三苯基膦为配体时产率为70%;相对而言,双膦配体能够提供更好的反应产率,其中以1,2-双(二苯基膦)乙烷(dppe)为配体时反应结果最佳,产率达到了85%(见表1中序号2).
接着,以dppe为配体对钯催化剂进行了筛选,包括四(三苯基膦)钯和醋酸钯都能得到反应产物,但还是以双(二亚芐基丙酮)钯的催化效率最高(见表1中序号2,6和7).
最后,对反应溶剂进行了研究.其中,醚类溶剂能够提供较高的产率,例如1,4-二氧六环同样以比较理想的产率(78%)得到了相应的烯丙基化产物,而甲苯和N,N-二甲基甲酰胺则产率都很低(见表1中序号2,8~10).

表1 α-苯乙酰基苯并三唑和烯丙基碳酸乙酯的Tsuji-Trost反应条件优化
注: 1)Pd(dba)2:双(二亚芐基丙酮)钯;Pd(PPh3)4:四(三苯基膦)钯;Pd(OAc)2:醋酸钯.
2)PPh3:三苯基膦;dppe:1,2-双(二苯基膦)乙烷;dppp:1,3-双(二苯基膦)丙烷;dppb:1,4-双(二苯基膦)丁烷;dppf:1,1′-双(二苯基膦)二茂铁.
3)分离产率.
2.2 反应底物的拓展
笔者运用以上“一锅法”烯丙基化反应的方法对不同取代的2-芳基-4-戊烯基酯的合成进行了研究.令人高兴的是,乙醇也可以代替甲醇进行酯化反应,酰基苯并三唑可以顺利地被置换得到相应的乙酯化合物3b,产率为82%(见表2).类似地,2-苯基-4-戊烯基异丙酯(3c)也可以被合成,但是与相应的甲酯、乙酯相比,其产率有明显的下降,只有65%(见表2).这说明第2步酯化反应时醇位阻的增加可能会不利于该反应的进行.

表2 不同取代的2-芳基-4-戊烯基酯的合成
另外,笔者对不同芳基取代的2-芳基-4-戊烯基酯的合成进行了研究.例如,氯原子和甲氧基在该反应条件下可以很好地被兼容,以良好的产率得到相应的2-芳基-4-戊烯基甲酯化合物(见表2:3d和3e).值得指出的是,氯原子和甲氧基可以通过偶联或脱保护等反应转变成其他取代基,为进一步合成官能团化的2-芳基-4-戊烯基甲酯化合物提供了可能.
化合物3a:产率80%,无色液体.1H NMR(600 MHz,CDCl3)δ:7.36~7.28(m,5H),5.75(ddt,J=17.1,10.2,6.8 Hz,1H),5.13~5.01(m,2H),3.69(s,3H),3.67(t,J=7.0 Hz,1H),2.89~2.82(m,1H),2.55(dt,J=14.2,7.4 Hz,1H);13C NMR(150 MHz,CDCl3)δ:173.9,138.5,135.2,128.6,127.9,127.3,117.0,52.0,51.4,37.6.
化合物3b:产率82%,无色液体.1H NMR(600 MHz,CDCl3)δ:7.38~7.28(m,5H),5.72~5.81(m,1H),5.12(d,J=17.1 Hz,1H),5.04(d,J=10.1 Hz,1H),4.21~4.10(m,2H),3.67(t,J=8.2 Hz,1H),2.87(dt,J=14.7,7.1 Hz,1H),2.55(dt,J=13.3,6.8 Hz,1H),1.24(t,J=7.1 Hz,3H);13C NMR(150 MHz,CDCl3)δ:173.4,138.6,135.3,128.5,127.9,127.2,116.9,60.7,51.5,37.6,14.1.
化合物3c:产率65%,无色液体.1H NMR(400 MHz,CDCl3)δ:7.34~7.25(m,5H),5.80~5.68(m,1H),5.13~4.96(m,3H),3.64~3.57(m,1H),2.87~2.78(m,1H),2.54~2.46(m,1H),1.23(d,J=6.2 Hz,3H),1.14(d,J=6.2 Hz,3H);13C NMR(100 MHz,CDCl3)δ:172.9,138.8,135.4,128.5,127.9,127.2,116.9,68.1,51.7,37.7,21.8,21.6.
化合物3d:产率70%,无色液体.1H NMR(400 MHz,CDCl3)δ:7.32~7.24(m,4H),5.76~5.63(m,1H),5.10~5.04(m,1H),5.02(d,J=10.2 Hz,1H),3.67(s,3H),3.63(t,J=7.6 Hz,1H),2.85~2.77(m,1H),2.50(dt,J=14.1,7.2 Hz,1H);13C NMR(100 MHz,CDCl3)δ:173.5,136.9,134.8,133.2,129.3,128.8,117.3,52.1,50.7,37.5.
化合物3e:产率70%,无色液体.1H NMR(600 MHz,CDCl3)δ:7.26(d,J=8.6 Hz,2H),6.89(d,J=8.6 Hz,2H),5.78~5.70(m,1H),5.10(d,J=17.1 Hz,1H),5.03(d,J=10.2 Hz,1H),3.82(s,3H),3.68(s,3H),3.63(t,J=7.8 Hz,1H),2.83(dt,J=15.0,7.2 Hz,1H),2.52(dt,J=13.9,7.1 Hz,1H);13C NMR(150 MHz,CDCl3)δ:174.1,158.8,135.3,130.6,128.9,116.9,114.0,55.2,51.9,50.5,37.6.
3 反应机理
笔者对反应可能存在的机理进行了探讨:烯丙基碳酸乙酯先与零价钯发生氧化反应形成π-烯丙基钯(见图4:1),苯乙酰苯并三唑作为亲核试剂进攻π-烯丙基钯形成中间体(见图4:2),最后在甲醇的亲核进攻下形成目标产物(见图4:3).

图4 可能的反应机理
4 结 论
本文发展了一个基于Tsuji-Trost反应的2-芳基-4-戊烯基酯化合物的“一锅法”合成方法.这是较为少见的钯催化硬亲核试剂的烯丙基化反应之一,是Tsuji-Trost反应的新发展.而且,该合成途径将Tsuji-Trost烯丙基化反应和酯化反应用“一锅法”进行,操作简单,无需中间体分离,减少了污染和能耗,是一种环境友好的合成方法.
[1]Martin P.Process for the preparation ofγ,δ-unsaturated esters and acids:US,4234741A[P].1980-11-18.
[2]Johnson W S,Werthemann L,Bartlett W R,et al.Simple stereoselective version of the Claisen rearrangement leading to trans-trisubstituted olefinic bonds.Synthesis of squalene[J].J Am Chem Soc,1970,92(3):741-743.
[3]Tsuji J.Palladium reagents and catalysts[M].Chichester:Wiley,2004:431-517.
[4]Katritzky A R,Wu Hong,Xie Linghong,et al.The origins of the benzotriazole project,its versatility illustrated by a new-C=CHCH+OEt synthon,and novel syntheses ofα,β-unsaturated aldehydes and ketones,furans,pyrroles and allyl ethers[J].Synthesis,1995,1995(10):1315-1323.
