作物遗传育种研究进展Ⅱ.作物有利基因鉴定和等位基因发掘
2014-08-07
(湖南农业大学,长沙410128)
作物遗传育种研究进展Ⅱ.作物有利基因鉴定和等位基因发掘
刘 忠 松
(湖南农业大学,长沙410128)
解析作物性状的遗传调控机制是提高育种选择准确性的基础,而鉴定控制性状的基因及其等位变异是解析作物性状的遗传结构的基础。简略介绍了基因鉴定的图位克隆方法、基于分离群体混合测序的鉴定方法、基于大量种质的基因组重测序GWAS方法和RNA测序方法,同时介绍了从种质资源中通过同源克隆方法发掘等位基因的方法,为科学有效利用最佳等位基因进行作物遗传改良提供依据。
作物;性状;遗传结构;基因鉴定;等位基因发掘
传统育种根据性状表现进行选择。生物任何性状都是基因型、环境及其互作的结果。同一基因型在不同环境下表现不同,不同基因型在某种环境条件下又可能有相同表现。因此导致依据表型进行育种选择不够准确。要提高育种选择的准确性,除了应严格控制试验环境外,对作物性状的遗传调控机制应有更好的了解,对选择方法应加以改进。
从遗传的角度来看,作物性状可分为质量性状和复杂性状(数量性状)。质量性状在群体中可以分为不同类型,但遗传简单,表现为少数基因控制,受环境影响小;复杂性状表现为连续变异,受多基因控制,受环境影响大。不管是哪种类型的性状,其形成都是生物生长发育过程基因表达的结果,实际上都涉及到多个基因。
为什么有些性状表现为简单遗传的质量性状,而其他性状表现为多基因控制的复杂性状,这与性状的遗传结构有关。如果控制某个性状的多个基因是按照反应顺序先后发挥作用,这种性状的遗传结构就是线性结构(图1a)。线性结构是遗传结构中最简单的类型,如果其中某个反应的基因突变丧失了功能,将导致整个链条崩溃,性状完全改变,如水稻半矮秆基因sd-1(即GA20OX2)突变,导致有活性的赤霉素不能合成,植株显著变矮。如果一个性状的形成不仅需要一些基因顺序发挥作用,而且还受到其他基因(如转录因子基因)的调控,这种调控基因不一定只调控一个反应,能调控多个甚至全部反应,这种性状的遗传结构可叫做分枝结构(图1b)。如果调控基因突变,将导致受其调控的基因表达改变,最终导致性状表达改变,如最近揭示的水稻新矮秆基因D53,在没有独角金内酯(Strigolactones)时抑制独角金内酯信号传导途径,但在有独角金内酯时,D53蛋白会被矮秆基因D3介导泛素化而降解,尽管D3、D14和D53突变体株高都表现为简单遗传,但D3和D14基因的状态影响D53基因作用的发挥。许多性状的形成不仅涉及到多个基因,而且还涉及多条途径,不同途径之间还存在交叉、互作,使得性状的遗传结构十分复杂,如同一张网络,这种性状遗传结构可叫做网络结构(图1c)。如水稻矮秆性状除了赤霉素合成和信号传导途径、独角金内酯合成和信号传导途径外,还发现d2、d11、d61和d62等矮秆突变与油菜素内酯合成和信号传导途径有关,另有数十个突变体形成矮秆的机制尚不清楚,水稻控制植株高度的网络结构尚未建立。具有网络遗传结构的性状,影响性状表达的基因作用有大有小,方向也并不完全一致,有些可能是正向的,有些可能是负向的。改良具有网络遗传结构的性状,鉴定控制性状形成的基因,发掘基因的等位变异是重要基础。
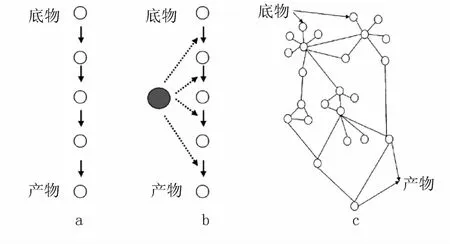
图1 性状遗传结构的类型
1 基因鉴定
连锁作图和关联作图都能初步定位控制性状的基因,但要最终鉴定控制性状基因的经典方法是图位克隆[1,2]。随着越来越多的作物有参考基因组序列,基因鉴定方法正在发生革命性的转变,利用新一代测序方法直接进行基因组(重)测序鉴定和转录组测序鉴定基因将会越来越流行。
1.1 图位克隆鉴定有利基因
图位克隆鉴定有利基因的基础是基因定位即找到与性状紧密连锁的标记。基因定位有连锁分析(家系分析)和关联分析(群体分析)两条途径。连锁分析需要构建性状分离的作图群体,对群体进行标记基因型分析和目标性状鉴定,找到目标性状两侧的标记,界定控制目标性状的基因组区域。为了加速这一过程,现多采用分离群体两极植株、F2代隐性植株和F1或近等基因系(NIL)进行分析。基因初步定位(到某一染色体)后,还需要开发新标记,利用大的分离群体(精细定位群体大小估算公式:N=Log(1P)/Log{1-[T-marker×3/λT]/100R},式中N为预测的群体大小;P为期望成功概率;T-marker是期望定位基因(两个标记之间)的精度(kb);λT为在两翼标记之间期望找到的重组单株数目;R为重组频率(kb/cM)[3]。如假设P=0.95、λT=9、T-marker=40 kb、R=250 kb/cM,则N=Log(1-0.95)/Log{1-[40×3/9]/25 000}=5 616)进行精细定位,缩小目标基因所在的基因组区域。单核苷酸多态性(SNP)芯片的开发和应用使得基因定位速度显著加快、分辨率显著提高。随着遗传标记的增加、加密,关联分析已越来越多地用来鉴定控制目标性状的基因。关联分析利用不同基因位点等位基因间的连锁不平衡(Linkage disequilibrium,LD)关系,进行标记与性状的相关性分析,以达到鉴定控制目的性状基因(或基因组区段)的目的。关联分析不需构建作图群体,而是利用自然形成的种质资源,分辨率更高,基因定位更精确,且可同时检测出一个基因的多个等位基因。但应注意种质资源的选择,防止因群体结构带来的假关联结果。
用精细定位的两侧标记或共分离标记筛选该作物的基因组文库(如BAC文库),构建BAC重叠群,重叠群的BAC测序,所得到的序列用于基因预测,预测的基因进行转基因遗传互补功能验证。有参考基因组序列的作物(如水稻)可利用精细定位标记直接确定目标基因候选区域,分析候选区域的基因,直接进行基因测序和比较,可鉴定出目的基因。
对于复杂性状,找到控制该性状的多个基因组区域(或数量性状位点,即QTL)后,需要从所有QTLs中选择贡献大的QTL,培育目标QTL的NIL,用含目标QTL的NIL与轮回亲本杂交培育分离群体,进行精细定位和后续步骤,也能克隆控制主效QTL的基因。
1.2 基因组(重)测序鉴定有利基因
近十多年,由于二代、三代基因组测序技术的迅猛发展,作物基因组测序也取得了前所未有的进步,继2002年完成水稻全基因组序列草图之后,高粱、玉米、大豆、马铃薯、粟、鹰嘴豆、木薯、芝麻、甜菜等作物以及普通小麦A和D基因组、四倍体棉花D基因组供体野生种的基因组序列[4]的基因组测序相继完成,为利用分离群体的单株或极端个体混合样本测序鉴定有利基因奠定了基础。
通过大量(100~1 000份)种质基因组重测序进行全基因组关联(GWAS)分析鉴定控制重要农艺性状的基因已在水稻、玉米和粟等作物上应用。GWAS分析能找到控制性状的候选基因[5~7]。
通过基因组测序鉴定有利基因的策略已开发出SHOREmap、NGM、MutMap和NIKS等几种方法。这些方法都是通过分离F2代群体混合高通量测序、测序结果与参考基因组序列连配鉴定SNP,根据SNP频率确定候选区域,进一步进行候选区域基因的功能分析验证。
SHOREmap法[8]利用大量(500株)F2代突变表型植株混合DNA进行深度(22倍)测序,测序得到的短序列利用短序列分析线SHORE与参考基因组序列连配,分析双亲等位基因的相对频率,用染色体作横坐标、等位基因相对频率作纵坐标绘图(SHOREmap区间图)。由于理论上与目标基因不连锁的基因来自双亲的等位基因频率相等,只有与目标基因紧密连锁的基因,其等位基因频率偏向亲本之一(突变体类型),染色体上这个等位基因偏向某个亲本的狭窄区域就是候选基因所在区域。将候选区域鉴定出的突变输入SHOREmap“注释功能”,将根据离等位基因分布峰的距离排列碱基替换、预测碱基改变的效应,找到候选区域、候选基因。
NGM法[9,10]利用F2代小群体(实用80株植株,推荐可用50株)混合DNA进行高通量深度测序,所得到的短序列利用Maq短序列作图软件包与参考基因组序列进行连配,鉴定出SNPs,并用统计值CHD鉴定SNP类型。如果CHD值接近0,说明测序结果与参考序列完全一致,该SNP位点为亲本纯合类型,如果CHD值接近0.5,说明该SNP位点为杂合类型,如果CHD值接近1,说明测序结果完全不同于参考序列,该SNP位点为突变纯合类型,只有CHD值为1的SNP代表真正的突变或连锁的突变位点。通过全基因组扫描缺乏CHD值为0.5、富集CHD值为1的染色体区段,并根据与参考基因组序列的差异程度鉴定候选基因区域,对候选区域的非同义SNP进行进一步验证。
MutMap法[11]是将野生型与纯合隐性突变体杂交,所得F1植株自交产生F2代,从大约20株隐性纯合的F2植株提取DNA混合,将混合DNA用新一代测序技术进行10倍或更高倍数覆盖的测序,通过序列连配分析突变体与野生型在单核苷酸多态性(SNP)、删除插入(InDel)等方面的差异。理论上,只有基因本身或其附近的SNP在100%的序列读段中表现出差异,而不是随机分布的50%。将突变体类型SNP的读段数目与总的读段数目之比叫做SNP指数,靠近目标基因的SNP指数应为1,而不连锁的SNP指数应为0.5,通过全基因组筛选,指数为1的一连串SNP所在区域理论上应包含目标基因,因为10倍覆盖随机出现SNP指数为1的概率为(0.5)10即10-3。在水稻连续4个以上SNPs具有SNP指数为1的概率≤2.3×10-9。找到一连串SNP指数为1的SNPs后,仔细检查其蛋白质编码区SNP是否发生了非同义替代。鉴定出候选基因之后,如同图位克隆鉴定一样,也需要进行遗传互补功能验证。对于不易发生重组的染色体区域如着丝粒区域,为了促进重组、交换,需要将F2植株自交获得大量F3代,用F3代进行分析。
NIKS法[12]直接测定野生型和突变体的基因组序列覆盖25倍以上,然后用Jellyfish软件计数各自样本测序序列中长度为k聚(k-mer,应短于测序读长,可采用多个不同k值,作者测序读长为33 nt,实际采用31 nt)序列的数目,绘制k-mer序列频率分布柱状图,鉴定样本特有的k-mer序列,将各样本的k-mer序列归并为种子序列,野生型和突变体的种子序列配对,其中一个代表野生等位基因,一个代表突变等位基因,然后选择与种子序列对至少共有一个k-mer的序列对进行组装,延长种子序列到数百bp长度(图2)。鉴定出突变位点后,注释突变对基因功能的影响。作者指出,杂合突变会影响种子序列对的鉴定,重复序列区域的突变不能用NIKS法鉴定,对于多态性密度高的材料应采用Cortex软件计数k-mer序列,为了减少需鉴定的突变数目,也可用突变体与野生型杂交F2代的混合DNA进行测序,或用2个以上不同等位基因的材料直接测序,然后找出同一基因中不同的突变。
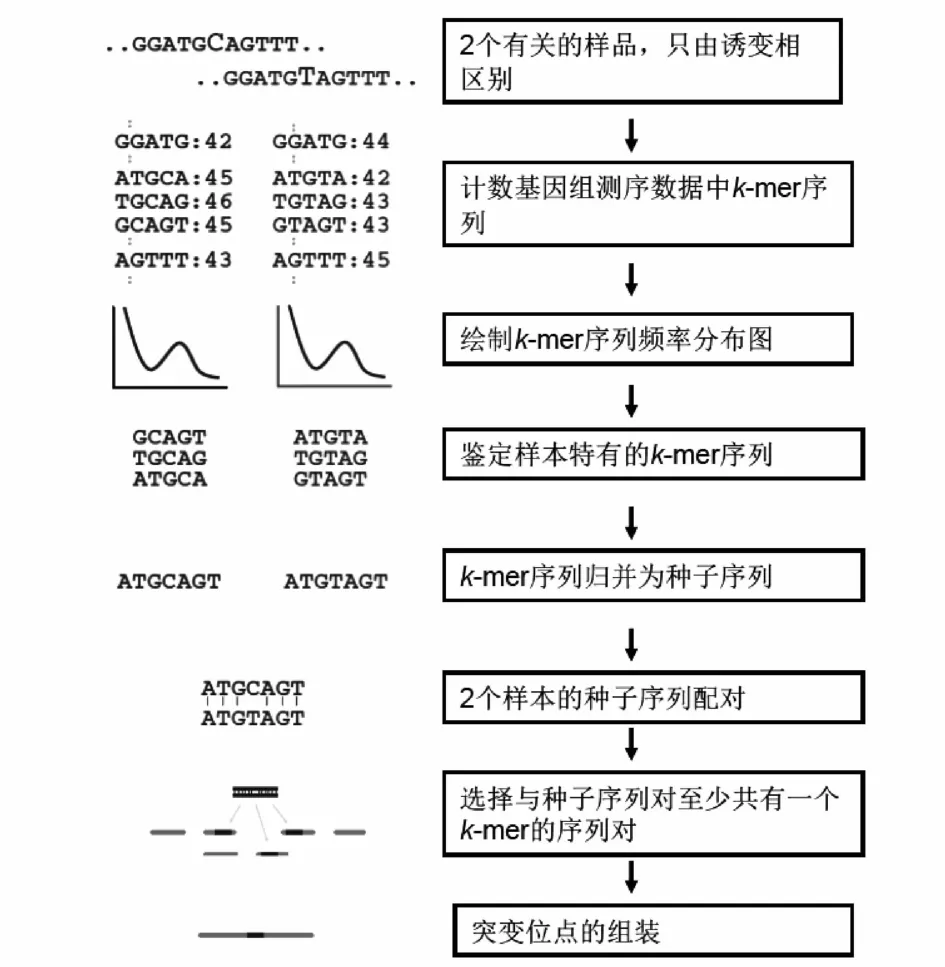
图2 NIKS法鉴定突变基因的基本程序
1.3 转录组测序鉴定有利基因
有人估测,人类已对玉米基因组的4%进行了选择。根据已注释基因估测玉米转录组只占基因组的4%。转录组测序更容易,在性状表达差异的材料中找到的差异表达基因有可能是控制该性状的基因。通过转录组测序不仅能鉴定出调控基因,而且能揭示该性状形成的途径。
转录组测序鉴定差异表达基因时,提取野生型和突变体性状表达组织的总RNA进行RNA深度测序(100倍),所得到的序列组装成unigenes,比较unigenes在野生型和突变体之间的表达量差异,结合KEGG分析、代谢产物分析等鉴定出候选关键基因。对于候选基因,可以利用转录组测序获得的序列设计引物进行扩增,比较基因组多态性和表达差异,验证候选基因的功能和影响的步骤[13]。如刘显军等通过转录组测序分析控制芥菜型油菜种皮颜色的基因[14]。
2 等位基因发掘
由于基因突变、基因重组、转座子转座等原因,生物不仅不断产生新的基因,而且原有基因可能形成新的变异类型(如碱基改变、序列插入或删除)即新的等位基因,使得一个基因可能有多个等位基因,如水稻“绿色革命”半矮秆基因sd-1已发现4个等位基因。不同等位基因的功能有时并不完全相同,如基因编码区的变异可能导致基因功能完全丧失,而基因调控区的改变可能只影响该基因的表达模式,因此育种上选择出最佳等位基因具有重要意义。
遗传学通过遗传互补试验确定基因是否等位,关联作图通过对自然群体的分析能鉴定出等位基因。随着越来越多的有利基因被鉴定、克隆,利用已克隆基因的序列,通过同源克隆挖掘种质资源中蕴藏的大量新等位基因已成为一种简便易行的方法。
同源克隆法发掘等位基因的基本程序见图3[15]。在等位基因发掘之前,需要确定目标性状,用最小的种质(核心种质)代表最大的遗传多样性,对拟用种质资源在适当环境下进行准确的性状鉴定,并且了解控制该性状的遗传结构、关键基因及其作用大小,根据前人研究结果选择目标基因,并查询同种作物或近缘植物有关该基因的序列。进行等位基因发掘时,设计扩增基因全长序列的引物,需要找到基因的保守区域,以便设计的引物适合在近缘植物中扩增出同源基因,不同引物对至少重叠100~200 bp,使扩增产物能覆盖基因编码区和上游调控区域,扩增产物测序应采用单扩增子,用性状高表达的种质的扩增子测序,建立同时包含多态性与表型数据、分类规范、连接方便的数据库。为了比较种质间同源基因序列的差异,可用ClustalW、BioEdit等软件进行分析。除了扩增产物测序外,TILLING分析也能检测等位基因的点突变(SNP多态性)。如Bhullar等采用同源克隆法从1 320份小麦种质中发现7个新的抗白粉病(Powdery mildew)Pm3等位基因[16]。
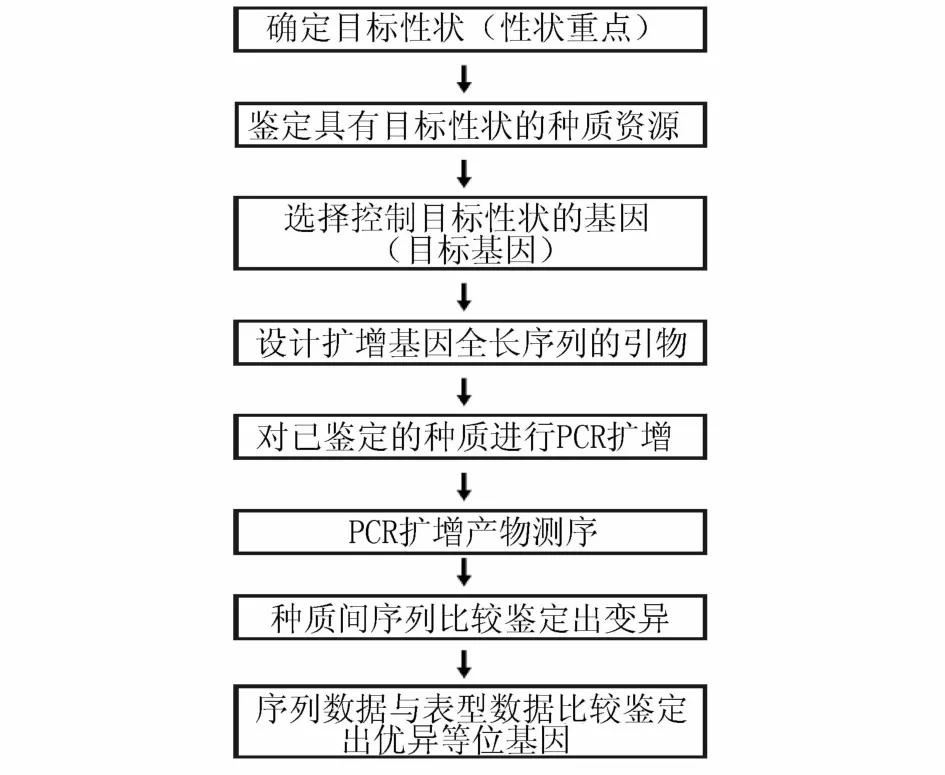
图3 同源克隆法发掘等位基因的基本程序
随着全基因组测序成本的降低,群体基因组学迅速发展,对大量种质资源进行重测序也正在成为挖掘种质资源中蕴藏的新等位基因的一条途径。
[1] Peters JL,Cnudde F,Gerats T.Forward genetics and map -based cloning approaches[J].Trends Plant Sci,2003,8:484-491.
[2] Lee J,Koh HJ.Gene identification using rice genome sequences[J].Genes Genom,2013,35:415-424.
[3] Dinka SJ,Campbell MA,Demers T,et al.Predicting the size of the progeny mapping population required to positionally clone a gene[J].Genetics,2007,176:2035-2054.
[4] Bolger ME,Weisshaar B,Scholz U,et al.Plant genome sequencing—applications for crop improvement[J]. Curr Opinion Biotech,2014,26:31-37.
[5] Han B,Huang X.Sequencing-based genome-wide association study in rice[J].Curr Opinion Plant Biol,2013,16:133-138.
[6] Huang X,Wei X,Sang T,et al.Genome-wide association studies of14 agronomic traits in rice land races[J]. Nat Genet,2010,42:961-967.
[7] Huang X,Zhao Y,Wei X,et al.Genome-wide associa-tion study of flowering time and grain yield traits in a worldwide collection of rice germplasm[J].Nat Genet,2012,44:32-39.
[8] Schneeberger K,Ossowski S,Lanz C,et al.SHOREmap:simultaneousmapping and mutation identification by deep sequencing[J].Nat Methods,2009,6:550-551.
[9] Austin RS,Vidaurre D,Stamatiou G,etal.Next-generation mapping of Arabidopsis genes[J].Plant J,2011,67:715-725.
[10]Austin RS,Chatfield SP,Desveaux D,etal.Next-generation mapping of genetic mutations using bulk population sequencing[J].Methods in Molecular Biology,2014,1062:301-315.
[11]Abe A,Kosugi S,Yoshida K,et al.Genome sequencing reveals agronomically important loci in rice using MutMap[J].Nat Biotechnol,2012,30:174-178.
[12]Nordström KJ,Albani MC,James GV,et al.Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers[J].Nat Biotechnol,2013,31:325-330.
[13]Hirsch CN,Buell CR.Tapping the promise of genomics in specieswith complex,non-model genomes[J].Annu Rev Plant Biol,2013,64:89-110.
[14]Liu XJ,Lu Y,Yuan Y,et al.De novo transcriptome of Brassica juncea seed coat and identification of genes for the biosynthesis of flavonoids[J].PLoS One,2013,8(8):71-110.
[15]Kumar GR,Sakthivel K,Sundaram RM,et al.Allelemining in crops:prospects and potentials[J].Biotechnol Adv,2010,28:451-61.
[16]Bhullar NK,Street K,Mackay M,et al.Unlocking wheat genetic resources for themolecular identification of previously undescribed functional alleles at the Pm3 resistance locus[J].PNAS,2009,106:9519-9524.
“作物镉污染防控技术研讨会”在长沙召开
简讯
镉(Cd),一种生物蓄积性强、毒性持久、具有“三致”作用的剧毒元素。摄入过量的镉对人体的危害极其严重。许多研究表明,我国部分地区的稻米镉含量超标(0.2 mg/kg)。如何消减稻米中镉的积累,现已成为保障粮食质量安全的一项首要科学任务。
2014年3月3日,湖南省作物学会组织相关领域的专家在湖南农业大学召开了“水稻镉污染防控技术研讨会”,来自全省的20多位专家教授出席会议。会上代表们讨论了辐污染防控技术,如阻断污染源、低镉型水稻品种的筛选与培育、降低土壤中镉的有效性、采用农艺措施调控水稻植株对镉的吸收与在籽粒中的积累等。代表们认为镉污染治理是一项世界性难题,需摸清其影响机理,采用协同研究攻关,做好长期探索的规划。(敖和军)
S330
:A
1001-5280(2014)02-0226-05
10.3969/j.issn.1001-5280.2014.02.28
2014 02- 21
刘忠松(1963-),男,湖南常宁人,博士,教授,从事作物遗传育种研究与教学。
