甲磺酸伊马替尼的合成工艺改进
2014-08-06徐晓霞史为龙南京优科生物医药有限公司江苏南京210018
徐晓霞,闵 涛,史为龙 (南京优科生物医药有限公司,江苏 南京 210018)
甲磺酸伊马替尼是瑞士Novartis公司研发的全球第一个根据肿瘤细胞活动原理设计的酪氨酸激酶抑制剂,经过广泛的临床验证后,被医学界誉为近年来有重大突破性的口服抗癌药物[1,2]。其化学名为4-(4-甲基-1-哌嗪)甲基-N-4-甲基-3-4-(3-吡啶)-2-嘧啶氨基苯基-苯胺甲磺酸盐。该药于2001年在美国首次上市,商品名为Gleevec,临床上用于治疗慢性粒细胞性白血病急变期、加速期或α-干扰素治疗失败后的慢性期患者,不能手术切除或发生转移的恶性胃肠道间质肿瘤(gastro-intestinal stromal tumours,GIST)患者。2002年2月,美国FDA批准该药用于治疗GIST患者[3,4]。
1 主要合成路线
目前,该药的合成路线众多,本研究旨在改进合成工艺,寻求适合工业化生产的工艺路线。有文献报道的该化合物合成路线主要有以下几种:
方法一[5]:以3-硝基苯基甲基胍和3-二甲氨基-1-(3-吡啶基)-2-丙烯1-酮为起始原料,经关环、还原、缩合得到伊马替尼(图1)。但反应路线和时间长,每步的收率低,不适合工业化生产。
方法二[6]:以N-(3-硝基苯基)-4-(3-吡啶基)-2-嘧啶胺为起始原料经氯化亚锡还原,再与4-氯甲基苯甲酰氯反应,最后与N-甲基哌嗪反应,生成伊马替尼。但该直链式的工艺成本较高(图2)。
方法三[7]:以4-甲基-3-硝基苯胺为起始原料,依次与对氯甲基苯甲酰氯和氮甲基哌嗪进行缩合反应,然后将硝基还原为氨基,再与单氰胺生成胍,最后与3-二甲氨基-1-(3-吡啶基)-2-丙烯-1-酮进行环合反应得到伊马替尼(图3)。其缺点是生成嘧啶环的环合反应收率很低,反应时间较长,且反应不完全。
方法四[7](图4):所用的酰胺缩合剂三甲基铝是易自燃的化学品,生产中存在潜在危险,不适合工业化生产。并且合成过程中要用昂贵的催化剂和BINAP配体,产品制备过程中产生异构化杂质,纯化困难。
众多路线中,以4-甲基-N3-[4-(3-吡啶基)嘧啶-2-基]-1,3苯二胺,4-[(4-甲基哌嗪-1-基)甲基]苯甲酸二盐酸盐为中间体的合成路线是最简便和快捷的,主要有两种方法:①用吡啶作碱和溶剂,4-[(4-甲基哌嗪-1-基)甲基]苯甲酸二盐酸盐与氯化亚砜反应合成酰氯盐酸盐,然后再将4-甲基-N3-[4-3-吡啶基)嘧啶-2-基]-1,3苯二胺加入到酰氯体系中反应,制备伊马替尼游离碱。②4-甲基-N3-[4-(3-吡啶基)嘧啶-2-基]-1,3苯二胺,4-[(4-甲基哌嗪-1-基)甲基]苯甲酸二盐酸盐用DCC和DMAP直接缩合,制备伊马替尼游离碱。第一种方法要用大量的吡啶,后处理需要处理的溶剂体系巨大,对操作人员的健康有较大伤害,环境污染严重,而且产物中的吡啶气味难以除去;第二种方法的后处理十分困难,由于采用了缩合剂DCC和催化剂DMAP,使得反应体系成分复杂,除尽DCC的转化产物DCU十分困难,加上产品和DCU的水溶性都很差,导致纯化难度高。

图1 伊马替尼合成路线(1)

图2 伊马替尼合成路线(2)
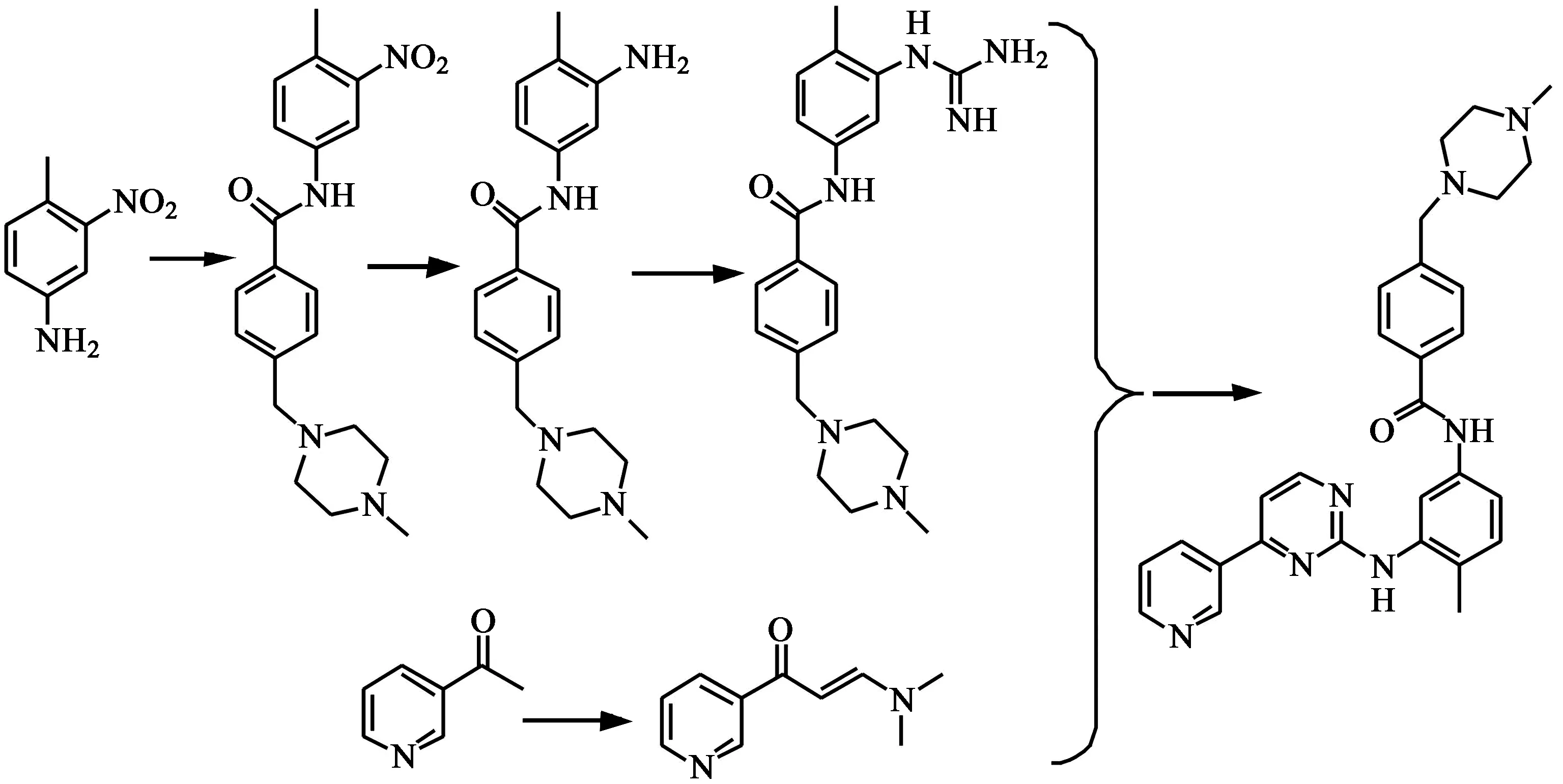
图3 伊马替尼合成路线(3)

图4 伊马替尼合成路线(4)
本实验经较长时间的反复摸索,最终确定第一步选用酰氯,使得该步的缩合获得很高收率,而且缩合反应采用水相反应,反应迅速,避免使用大量有机溶剂,因此,整条工艺链的污染量很小,操作环境好,避免伤害操作人员,同时产品纯度高,十分适合工业化生产。

图5 甲磺酸伊马替尼的合成路线
2 实验方法
2.1仪器与试剂 4-甲基-N3-[4-(3-吡啶基)嘧啶-2-基]-1,3苯二胺,工业级;4-[(4-甲基哌嗪-1-基)甲基]苯甲酸二盐酸盐,工业级;乙醇,工业级;甲磺酸,工业级。熔点仪:YRT-3熔点仪。HPLC测试方法:十八烷基硅烷键合硅胶为填充剂(Waters symmetry,5 μm, 3.9 mm×150 mm),检测波长267 nm,柱温40℃。取辛烷磺酸钠7.5 g,加水1 000 ml溶解,用10%磷酸调节pH值至2.5,为流动相A;甲醇为流动相B。流速1.2 ml/min。最初的15 min,42%流动相A、58%流动相B;然后开始线性增加流动相B的比例,至25 min时,流动相B的比例为87%;再减少流动相B的比例,至30 min时,流动相B的比例为58%,保持10 min,柱效应不低于3 000。
2.24-[(4-甲基-1-哌嗪)甲基]-N-[4-甲基-3-[4-(3-吡啶)-2-嘧啶]氨基-苯基]-苯甲酰胺(伊马替尼游离碱)的合成 将4-[(4-甲基哌嗪-1-基)甲基]苯甲酸二盐酸盐(100 g,0.37 mol)投入容量为1 L的三口瓶,加入氯化亚砜500 ml,N,N′-二甲基甲酰胺(DMF)4 ml,搅拌升温,回流反应8 h,降至室温,抽滤,正己烷(100 ml×2)洗涤,25~30℃真空干燥3 h,得白色固体酰氯盐酸盐100.3 g。
将4-甲基-N3-[4-(3-吡啶基)嘧啶-2-基]-1,3苯二胺(45 g,0.16 mol)投入容量为1 L的三口瓶,加入750 ml纯化水,搅拌内温降至0~3℃,分批加入上步所得的酰氯盐酸盐,加料后继续搅拌反应1 h,抽滤,滤液采用纯化水(45 ml×2)洗涤,洗涤的滤液中加入丙酮375 ml,搅拌升温至内温50℃,用25%氨水将pH值调至8~9,降内温至20~25℃,搅拌析晶3 h,抽滤,滤饼用少量冰丙酮洗涤,45~50℃真空干燥,得到伊马替尼游离碱73 g,收率91.2%,熔点(mp):212~213℃(文献211~213℃)。HPLC纯度为99.4%。1H NMR(DMSO-d6):10.15(s,1H,-NHCO-),9.27(s,1H),8.95(s,1H),8.66(dd,1H),8.51(dd,1H),8.46~8.48(m,1H),8.07(s,1H),7.90(dd,2H),7.42~7.53(m,5H),7.20(dd,1H),3.52(s,2H),2.36~2.50(m,8H),2.22(s,3H),2.15(s,3H)。ESI-MS: 494.2。
2.3甲磺酸伊马替尼的合成 将伊马替尼游离碱73 g悬浮于300 ml乙醇中,加热回流,然后滴加甲磺酸(14.2 g,0.15 mol)的乙醇溶液,搅拌反应2 h,降温到10~20℃析晶8 h,抽滤,乙醇洗涤,真空干燥,得到类白色固体78 g,收率为89.6%,mp:213~215℃,HPLC纯度≥99.7%。1H NMR(DMSO-d6):0.18(s,1H,-NHCO-),9.4(s,1H,-NH-),9.28(s,1H,CH),8.95(s,1H,OH),8.69(d,1H,CH),8.51(d,1H,CH),8.48(d,1H,CH),8.10(s,1H,CH),7.96(d,1H,CH),7.53(s,1H,CH),7.52(s,1H,CH),7.46~7.48(m,2H,CH×2),7.42(d,1H,CH),7.21(d,2H,CH×2),3.68(s,2H),3.38(s,2H),3.04(s,2H),2.93(s,2H),2.79(s,3H),2.37(s,5H),2.23(s,3H)。ESI-MS[M+H]+: 590.7。
3 结果与讨论
选用4-甲基-N3-[4-(3-吡啶基)嘧啶-2-基]-1,3苯二胺和4-[(4-甲基哌嗪-1-基)甲基]苯甲酰氯盐酸盐,在较低的温度下,水相介质中实现缩合,可以简便地合成伊马替尼,反应程度高,产品纯度好,且收率与用吡啶作溶剂相当,环境污染少,可实现工业化生产。
【参考文献】
[1] 陈 敖,黄荷香,宋帅娟. 甲磺酸伊马替尼的合成[J]. 精细与专用化学品,2007,15(8):23-25.
[2] 赵 瑾,姜 达. 甲磺酸伊马替尼耐药机制及其逆转研究进展[J]. 癌症进展杂志,2009,7(2):158-165.
[3] 陈灵娟,周和平,黄文峰,等. 甲磺酸伊马替尼的合成[J]. 河北科技大学学报,2011,32(5):492-495.
[4] 王保国,刘玉凤,狄国杰. 甲磺酸伊马替尼的合成[J]. 济宁医学院学报,2012,35(2):103-105.
[5] 齐默尔曼 J. 嘧啶衍生物及其制备方法[P]. 中国专利,93103566,1993-10-27.
[6] Kompella A,Bhujanga RA,Venkaiah CN,etal. Process for the preparation of the anti-cancer drug imatinib and its analogues[P]. 世界专利,2004108699,2004-12-16.
[7] 卢瓦瑟勒尔, 考夫曼, 阿贝尔,等. N-苯基-2-嘧啶胺衍生物[P]. 中国专利,03803556,2003-02-06.
