不对称合成新型的吲哚螺-2,3-二氢呋喃衍生物*
2014-08-06邹建锋李新生
邹建锋, 李新生
(浙江师范大学 化学与生命科学学院,浙江 金华 321004)
二氢呋喃化合物是一类具有重要合成价值的环状烯醇醚,该结构单元的天然产物往往表现出一定的生物活性[1-3].近年来发现有的氢化呋喃类衍生物具有抗病毒、抗肿瘤的效用[4],由此对这类化合物的合成研究引起了有机化学工作者的关注.早期人们尝试利用炔醇环化异构化合成二氢呋喃类化合物,但这些方法往往涉及多步反应[5],比较繁琐,所得最终产物的产率也较低.
螺环化合物具有刚性结构,结构稳定,在医药、不对称催化、发光材料、防火材料、农药、高分子黏合剂等方面有重要的应用.近年来,螺环化合物在生物学及医学领域得到广泛应用,许多螺环化合物是医药和农药的有机中间体[6-9].同时,靛红等一系列羰基化合物被发现可以取代芳香醛在多组分反应中的地位,并已合成了大量含有螺环结构的杂环化合物[10].而且,螺环化合物一直是化学工作者热衷于研究的对象.
本文在最近报道的有关二氢呋喃类化合物的合成[11-12]及靛红类物质参与反应[13-15]的文献基础上,主要利用迈克(Michael)加成方法,在手性碱的作用下合成了一系列手性吲哚螺-2,3-二氢化呋喃衍生物(见图1),大大提高了二氢呋喃类化合物合成的产率.而且,到目前为止尚没有对此类二氢呋喃螺环化合物合成的报道.本实验得到了一些新化合物,产率均较高,且产物的结构用红外光谱、核磁共振氢谱、核磁共振碳谱和高分辨质谱进行了表证.

R= 1a:C6H5;1b:2-furanyl;1c:4-CH3O-C6H4;1d:4-Br-C6H4;1e:4-Cl-C6H4;1f:4-F-C6H4;1g:3-CH3-C6H4
1h:3-CH3O-C6H4;1i:3-Br-C6H4;1j:3-Cl-C6H4;1k:2-Br-C6H4;1l:2-Cl-C6H4;1m:4-Br-2-Cl-C6H3
图1 研究内容
1 实验部分
1.1 仪器和试剂
熔点用X-4 数字显微熔点测定仪(温度计读数未校正,上海精密科学仪器有限公司)测定;红外光谱(IR)用FTIR-8300PCS 红外光谱仪(KBr 压片)测定;核磁共振谱(NMR)用Bruker公司400MHZ 型核磁共振仪测定,氘代二甲基亚砜(DMSO-d6)为溶剂,四甲基硅烷(TMS)为内标;高分辨质谱(HRMS)用Bruker公司ESI型高分辨质谱仪测定;手性用岛津LC-20AT 高效液相色谱仪测定,Chiralcel AD-H,Chiralcel OD-H手性色谱柱;比旋光度用Perkin Elmer 343型自动旋光仪测定.
其他药品除特别说明外均为市售分析纯试剂.
1.2 吲哚螺-2,3-二氢呋喃衍生物的合成
在反应管中加入0.15 mmolN-甲基-3-羟基-2-吲哚酮(见图1:2),0.18 mmol氰基苯乙烯衍生物(见图1:1a~1m),0.007 5 mmol辛可宁及少量的0.4 nm分子筛,置于15 ℃环境中,再加入2 mL 1,3,5-三甲苯,搅拌均匀.薄层层析跟踪反应进程,60 h后完全反应.反应停止后进行柱色谱分离提纯,用100~150目硅胶柱,V(石油醚)∶V(乙酸乙酯)=2∶1洗脱.得到的纯产物干燥后用IR,1H NMR,13C NMR和HRMS进行表征.
2 结果与讨论
2.1 手性催化剂的筛选
首先,本实验选用图1中的1a与2作为反应底物,加入与底物1摩尔比为10%的不同的手性催化剂(见图2),以二氯甲烷为溶剂,在15 ℃下考察了该反应的情况,结果如表1所示.表1表明,这些催化剂都能催化该反应体系,并能给出很好的反应活性,但是普遍的对映选择性比较差.由表1可见,辛可宁(催化剂(3))可以相对较好地催化反应,并得到54%的ee(对映体过量百分数)值和90%的产率.所以确定辛可宁为本实验理想的手性催化剂.
2.2 不同溶剂的筛选
手性催化剂筛选后,以与底物1摩尔比为10%的辛可宁为催化剂,接着考察了不同溶剂对反应的影响.表2表明:二甲基亚砜(DMSO)和二甲基甲酰胺(DMF)对该反应给出了几乎不反应的结果;乙腈(CH3CN)、丙酮(CH3COCH3)、四氢呋喃(THF)和乙醇(CH3CH2OH)对该反应给出了比较低的反应活性;其他溶剂则表现出一定的反应活性;只有甲苯系列的溶剂给出了较好的反应活性,且比较之下1,3,5-三甲苯为反应的最佳溶剂,得到了70%的ee值和几乎是定量反应的产率(99%).

图2 手性催化剂
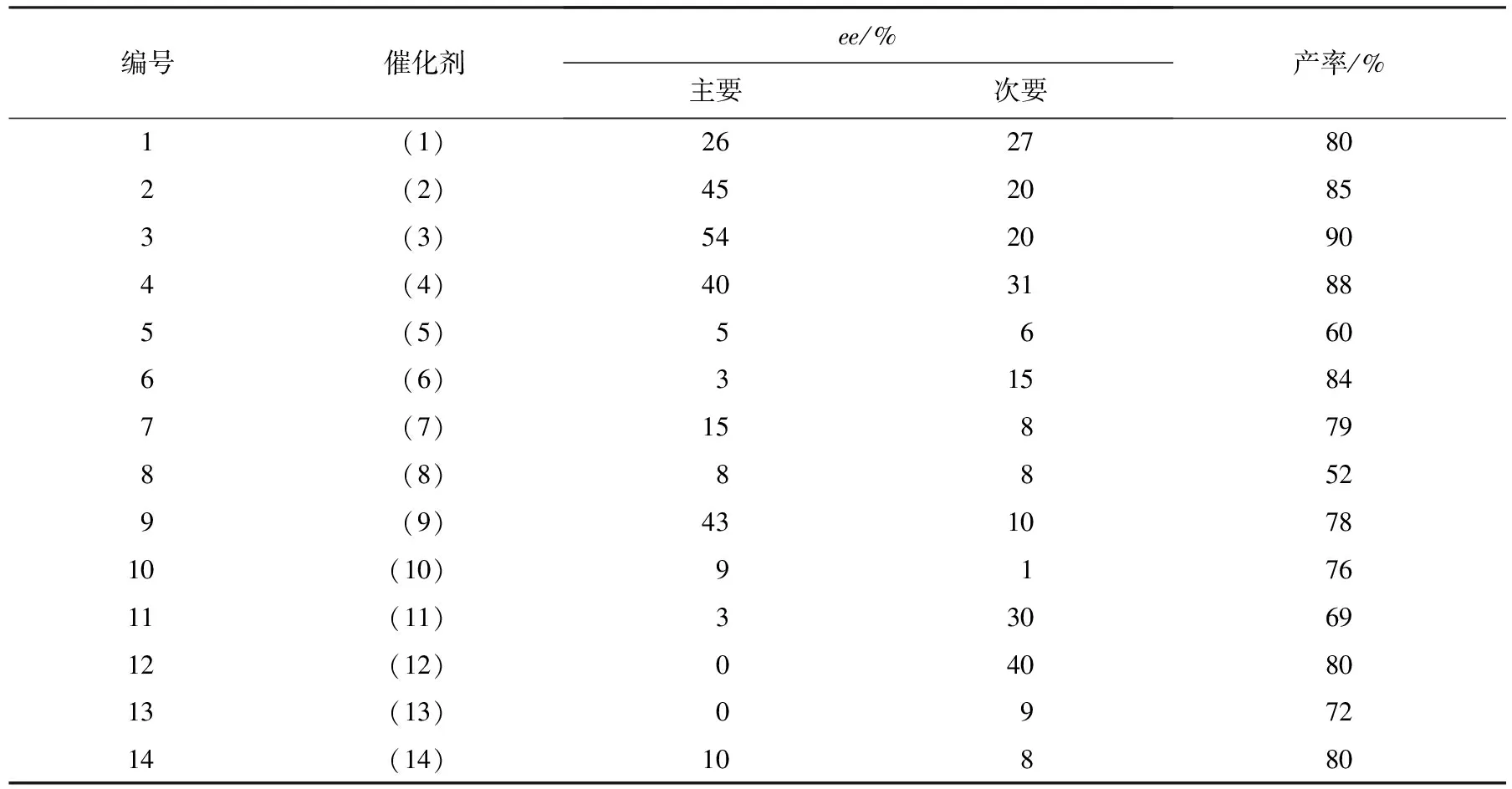
表1 筛选不同的手性催化剂(见图2)1)
注:1)溶剂为二氯甲烷;催化剂与底物1的摩尔比为10%.
2.3 反应温度和催化剂用量的筛选
在已经确定了手性催化剂和反应溶剂的基础上,本实验对不同的反应温度和催化剂用量进行了筛选.结果表明:15 ℃是最佳反应温度(见表3:编号4);当反应温度升高时,反应速率会大大加快,而选择性会降低;当反应温度降低时,反应速率和选择性都会降低.对催化剂用量进行筛选时发现:适当降低催化剂的用量至与底物1的摩尔比为5%时(见表3:编号7 ),同样可以得到产率99%的效果;而加大催化剂用量时,反应的选择性得不到明显的提高,反而有所降低.所以,本实验将催化剂与底物1的摩尔比为5%作为最佳的催化剂用量.本实验还发现,向反应体系中加入少量的分子筛可以提高反应的选择性(见表3:编号11).
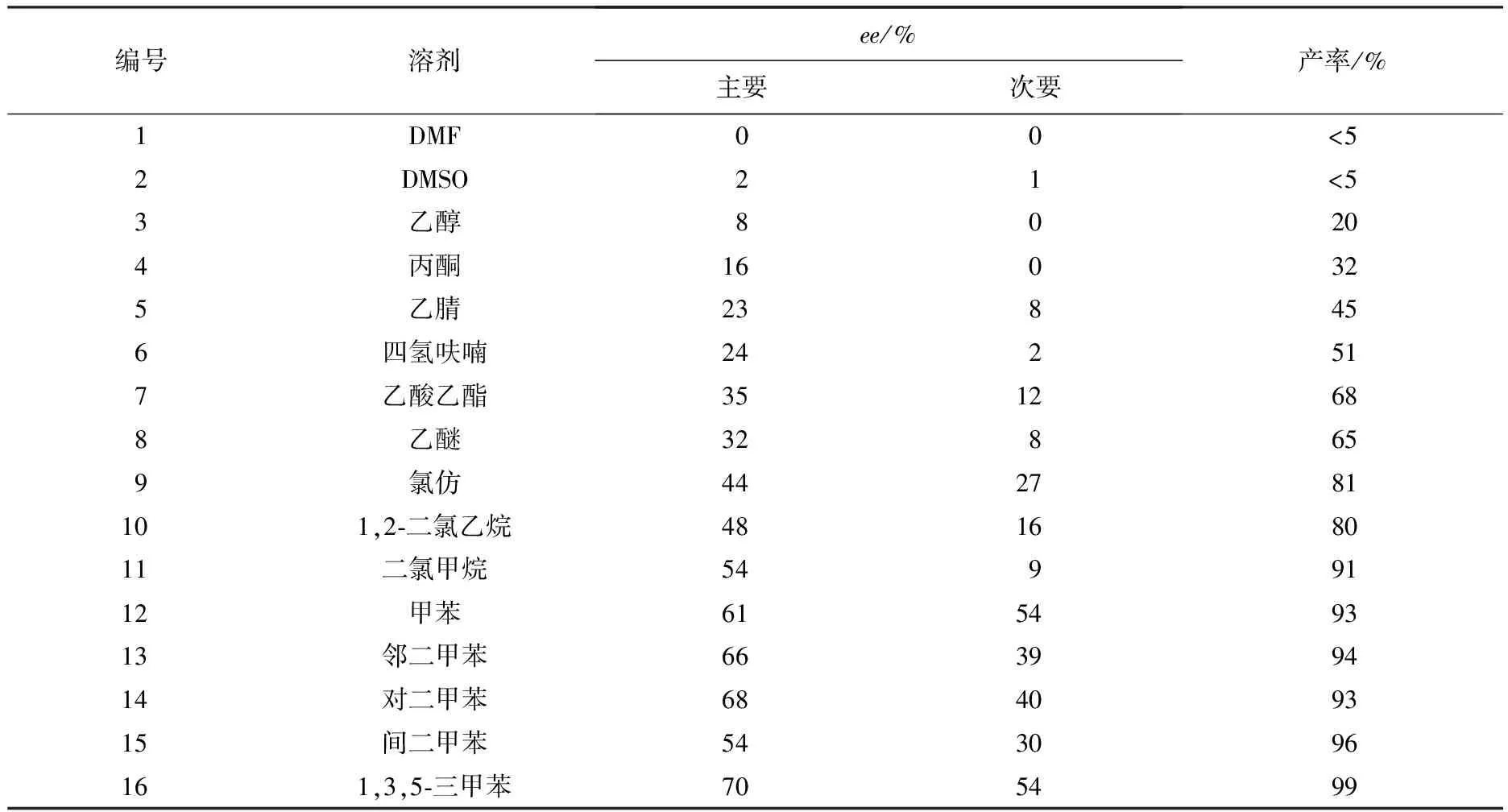
表2 筛选不同的反应溶剂1)
注:1)辛可宁与底物1的摩尔比为10%;产率由柱色谱分离测得.

表3 筛选反应温度及催化剂用量1)
注:1)产率由柱色谱分离测得.
最后,本实验确定该反应体系的最佳条件为:催化剂为与底物1摩尔比5%的辛可宁(cinchonine),1,3,5-三甲苯为溶剂,加入少量的0.4 nm分子筛,反应温度为15 ℃.本实验还跟踪了该反应并确定了反应的大致时间,60 h后反应完全.
2.4 底物扩展
为了验证以上筛选出来的条件是否对合成此类二氢呋喃化合物具有较好的催化活性,本实验选取了一系列在芳环上具有不同电子效应和位阻效应取代基的底物1(见图1),在最佳条件下进行了研究,结果见表4.与1a相比较,糠醛(1b)表现出比1a更好的选择性(见表4:编号2).取代基处于对位上,无论是供电子基团还是吸电子基团,其对映选择性与1a相比较变化都不大.间位的吸电子基团明显地降低了反应的对映选择性(见表4:编号9~10);而有供电子基团时,相比较1a而言,对映选择性变化也不大(见表4:编号7~8).有邻位取代基的芳香醛给出了稍低于1a的对映选择性(见表4:编号11~12).本实验发现,该反应条件下所有的底物1都具有很好的反应能力,产率都达到了90%以上.同时,本实验也对所有反应的非对映选择性进行了测定.
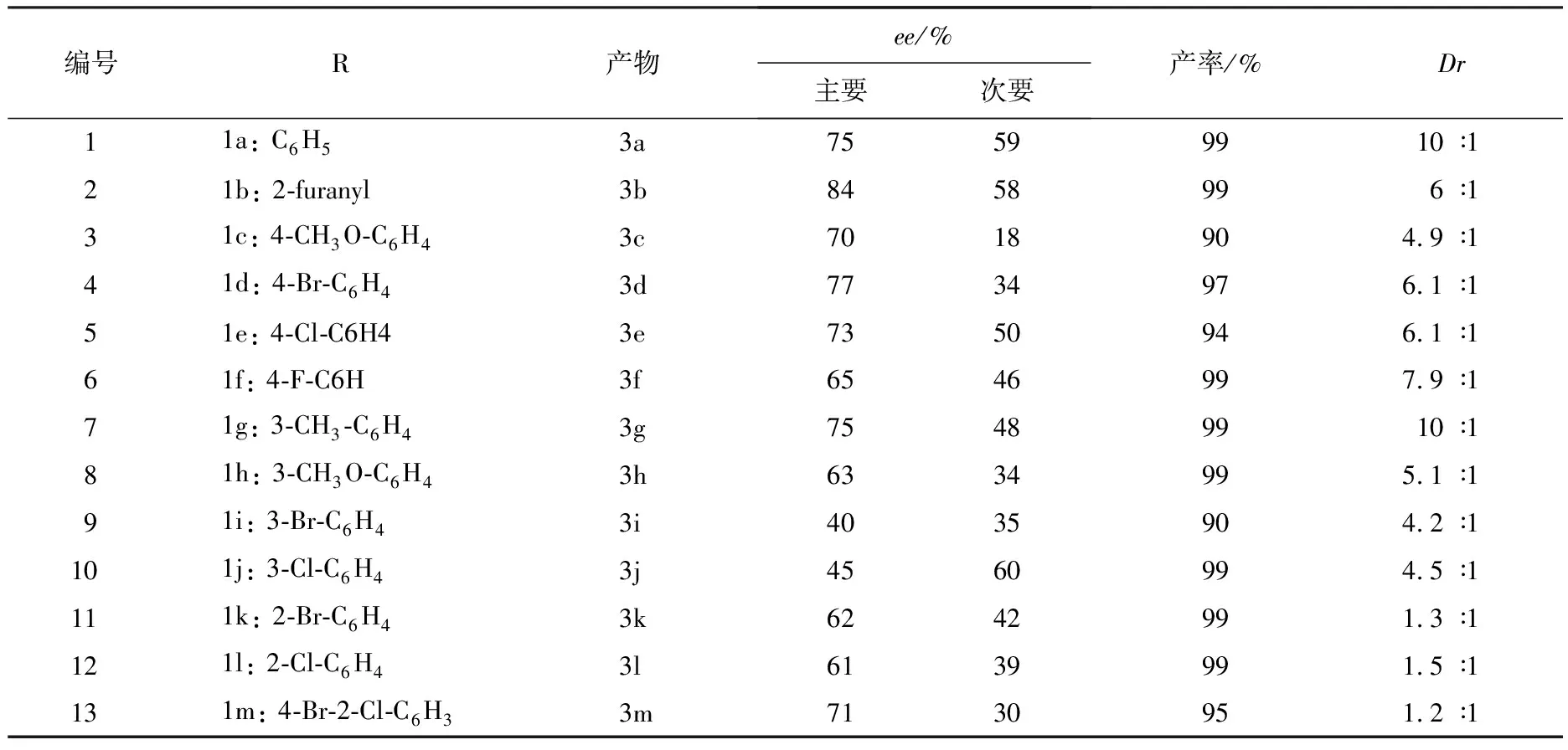
表4 所有底物1(见图1)在最佳条件下的反应1)
注:1)产率由柱色谱分离测得;Dr值由核磁共振氢谱仪测得.
2.5 化合物的结构表征













2.6 推断可能的反应机理
该反应主要是利用迈克加成发生的,在手性碱的作用下发生3,4-加成反应,再由羟基氧上的孤对电子进攻氰基上的碳原子,从而形成电子的转移,成功地构建了新型的吲哚螺-2,3-二氢呋喃类衍生物.可能的机理如图3所示.

图3 反应历程
3 结 论
本文主要研究了靛红衍生物(N-甲基-3-羟基-2-吲哚酮)与氰基苯乙烯类衍生物通过迈克加成方法,在辛可宁的催化下合成了一系列新型的手性吲哚螺-2,3-二氢呋喃类衍生物,合成产率都较高,也有较好的选择性,且后处理方便,为吲哚螺-2,3-二氢呋喃类衍生物的合成提供了新方法.
参考文献:
[1]Nakamura I,Chan C S,Araki T,et al.Synthesis of multisubstituted 2-(dihydrofuran-2(3H)-ylidene)acetates via intramolecular carboalkoxylation by platinum-olefin catalyst system[J].Org Lett,2008,10(2):309-312.
[2]Li Mengru,Lin Shaoxia,Dong Zhiyong,et al.Organocatalyzed anion relay leading to functionalized 2,3-dihydrofurans[J].Org Lett,2013,15(15):3978-3981.
[3]Chen Guodong,Cao Weiguo,Chen Jie,et al.High stereoselective synthesis oftrans-2,3-dihydrofuran derivatives[J].Synth Comm,2004,34(20):3793-3799.
[4]Sugimura H,Osumi K,Kodaka Y.Stereoselective synthesis of 2′-deoxy-β-D-threo-pentofuranosyl nucleosides by the NBS promoted coupling reaction of thioglycosides with silylated heterocyclic bases[J].J Org Chem,1994,59(25):7653-7660.
[5]Gianturco M A,Friedel P,Flanagan V.A novel procedure for the synthesis of 2,3-dihydrofurans[J].Tetrahedron Lett,1965,6(23):1847-1852.
[6] 魏荣宝,刘博,刘洋,等.具有生理活性的含氧、氮、硫杂原子螺环化合物的研究进展[J].有机化学,2008,28(9):1501-1514.
[7] 魏荣宝,刘洋,梁娅.含螺环结构农药研究进展[J].有机化学,2009,29(3):476-487.
[8]Gianotti M,Andreltti D,Casotto D,et al.Asymmetric route to spirotetracyclic (1S)-5′,11′-dihydro-3H-spiro[cyclopentane-1,10′-dibenzo[a,d]cyclohepten]-3-one derivatives[J].Tetrahedron Lett,2011,52(2):329-331.
[9] Chiba S,Zhang L,Lee J Y.Copper-catalyzed synthesis of azaspirocyclohexadienones fromα-azido-N-arylamides under an oxygen atmosphere[J].J Am Chem Soc,2010,132(21):7266-7267.
[10]Singh G S,Desta Z Y.Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks[J].Chem Rev,2012,112(11):6104-6155.
[11]Du Ding,Hu Zhongyuan,Tang Weifang,et al.A convenient synthesis of polysubstituted 2-amino-4,5-dihydrofuran-3-nitriles from benzoins or benzaldehydes[J].Tetrahedron Lett,2012,53(4):453-457.
[12]Alizadeh A,Rezvanian A,Zhu Longguan.Synthesis of heterocyclic [3.3.3] propellanes via a sequential four-component reaction[J].J Org Chem,2012,77(9):4385-4390.
[13]Bergonzini G,Melchiorre P.Dioxindole in asymmetric catalytic synthesis:routes to enantioenriched 3-substituted 3-hydroxyoxindoles and the preparation of maremycin A [J].Angew Chem Int Ed,2012,51(4):971-974.
[14]Trost B M,Hirano K.Dinuclear zinc catalyzed asymmetric spirannulation reaction:an umpolung strategy for formation ɑ-alkylated-ɑ-hydroxyoxindoles[J].Org Lett,2012,14(10):2446-2449.
[15]Retini M,Bergonzini G,Melchiorre P.Dioxindole in asymmetric catalytic synthesis:direct access to 3-substituted 3-hydroxy-2-oxindoles via 1,4-additions to nitroalkenes[J].Chem Commun,2012,48(27):3336-3338.
