6-羟基脱氧Canagliflozin的合成及其生物活性
2014-08-05天津中医药大学研究生院天津300193天津药物研究院天津市新药设计与发现重点实验室天津300193山东大学化学与化工学院山东济南250100
(1.天津中医药大学研究生院,天津 300193;2.天津药物研究院天津市新药设计与发现重点实验室,天津 300193;3.山东大学化学与化工学院,山东济南 250100)
(1.天津中医药大学研究生院,天津 300193;2.天津药物研究院天津市新药设计与发现重点实验室,天津 300193;3.山东大学化学与化工学院,山东济南 250100)
以Canagliflozin(1)为起始原料,经7步反应合成了其6-OH脱氧产物4-(6-脱氧-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(8),8是与母体1相似的新型SGLT2抑制剂,总收率21%,其结构经1H NMR,13C NMR和MS表征。体外生物活性测试结果显示,1和8对hSGLT2的IC50分别为3.9 nM和4.8 nM,对SGLT1的选择性(SGLT2/SGLT1)分别为178和194。在大鼠尿糖排泄实验(UGE)中8具有很强的尿糖排泄作用,与母体1并无显著性差异。
SGLT2抑制剂;Canagliflozin;脱氧;合成;抑制活性;尿糖排泄实验(UGE)
在糖尿病治疗中,血液流经肾小球时被过滤到原尿中的葡萄糖在肾近端小管中会被重新吸收到血液中,在这一重吸收过程中起主要作用的是Na+-依赖性葡萄糖共转运体2(SGLT2),它对重吸收的贡献约 90%,另外 10%的贡献来源于SGLT1[1-2]。因此抑制SGLT2就能增加尿糖排出,从而达到降低血糖的目的。SGLT2已成为近年来糖尿病治疗领域最有前景的新靶点,其中研究进展较快的SGLT2抑制剂Dapagliflozin(Chart 1)[3]和Canagliflozin(1)[4]已分别于2012年在欧盟和2013年在美国上市。
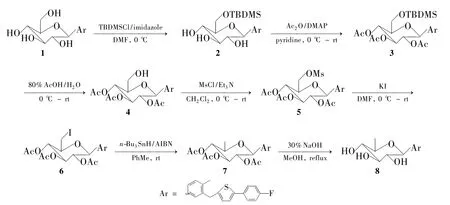
Scheme 1

Chart 1
在前期研究[5]中我们发现Dapagliflozin的6-OH脱氧产物具有比母体更强的抑制SGLT2的能力。为了研究该类6-OH脱氧引起活性增强的普遍性,本文合成了1的6-OH脱氧产物——4-(6-脱氧-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(8)。即1在咪唑作用下与叔丁基二甲基氯硅烷(TBDMSCl)反应,选择性保护葡萄糖环上的6-OH[6]得4-(6-O-叔丁基二甲基硅基-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(2);2在 4-二甲氨基吡啶(DMAP)催化下经Ac2O乙酰化,保护其葡萄糖环上的其余三个OH得4-(2,3,4-三-O-乙酰基-6-O-叔丁基二甲基硅基-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(3);3在80%醋酸水溶液中于40℃脱去TBDMS保护基得4-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(4);在Et3N作用下,4与甲磺酰氯(MsCl)反应,将其暴露的6-OH转化为甲磺酸酯得4-(2,3,4-三-O-乙酰基-6-O-甲磺酰基-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(5);5与KI于80℃反应,将甲磺酸酯转化为碘化物4-(2,3,4-三-O-乙酰基-6-脱氧-6-碘-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(6);用三正丁基氢化锡(n-Bu3SnH)和偶氮二异丁腈(AIBN)于室温还原6脱碘得4-(2,3,4-三-O-乙酰基-6-脱氧-β-D-吡喃葡萄糖基)-2-[5-(4-氟苯基)噻吩-2-甲基]-1-甲基苯(7);7在MeOH中用30%NaOH溶液水解脱去乙酰基得目标产物8(Scheme 1),其结构经1H NMR,13C NMR,ESI-MS和MS表征。并对1和8的生物活性进行了研究。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪(温度未校正);Bruker AV 400型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agilent Q-TOF 6510型质谱仪(电喷雾离子源,直接进样)。
1按文献[4]方法合成;其余所用试剂均为分析纯,其中DMF使用前经干燥处理。
1.2 合成
(1)2的合成
在反应瓶中依次加入DMF 50 mL,1 8.54 g (19.21 mmol)和咪唑3.93 g(57.68 mmol),搅拌使其溶解;冰水浴冷却,缓慢滴加TBDMSCl(4.64 g)的DMF(10 mL)和CH2Cl2(1 mL)溶液,滴毕,于室温反应过夜(TLC监测)。小心倒入300 mL冰水中,用CH2Cl2(3×100 mL)萃取,合并有机相,用饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析[洗脱剂:A=V (乙酸乙酯)∶V(石油醚)=3∶1]纯化得白色泡沫状固体 2 10.44 g,收率 98%;1H NMR δ: 7.54~7.58(m,2H),7.25(d,J=3.6 Hz,1H),7.16~7.20(m,3H),7.10(s,2H),6.76(d,J= 3.6 Hz,1H),4.89~4.90(m,2H),4.70(d,J= 6.0 Hz,1H),4.10(s,2H),3.96(d,J=9.6 Hz,1H),3.85(d,J=10.8 Hz,1H),3.18~3.26(m,3H),3.10~3.12(m,1H),2.25(s,3H),0.79 (s,9H),-0.04(s,3H),-0.08(s,3H)。
(2)3的合成
在反应瓶中依次加入吡啶50 mL和2 10.00 g(17.90 mmol),搅拌使其溶解;冰水浴冷却,缓慢滴加Ac2O 30 mL,滴毕,加入DMAP 1.00 g,于室温反应过夜(TLC监测)。小心倒入300 mL冰水中,用CH2Cl2(3×100 mL)萃取,合并有机相,依次用3%盐酸(2×100 mL)和饱和食盐水(2× 100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂:A=1∶5)纯化得白色固体3 11.60 g,收率94%,m.p.94℃~96℃;1H NMR δ:7.54~7.58(m,2H),7.26(d,1H,J=3.6 Hz),7.11~7.21(m,5H),6.74(d,J=3.6 Hz,1H),5.30(t,J=9.4 Hz,1H),5.08(t,J=9.6 Hz,1H),4.87(t,J=9.8 Hz,1H),4.58(d,J= 9.6 Hz,1H),4.06~4.15(m,2H),3.82(dd,J= 2.4 Hz,10.0 Hz,1H),3.69~3.71(m,1H),3.63(dd,J=4.4 Hz,11.6 Hz,1H),2.25(s,3H),1.99(s,3H),1.90(s,3H),1.71(s,3H),0.81(s,9H),-0.02(s,3H),-0.08(s,3H)。
(3)4的合成
在反应瓶中依次加入冰醋酸80 mL和3 11.50 g(16.79 mmol),搅拌使其溶解;缓慢滴加水20 mL,于40℃反应约5 h(TLC监测)。倒入300 mL冰水中,用CH2Cl2(3×100 mL)萃取,合并有机相,依次用饱和 NaHCO3溶液(2×100 mL)和饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂: A=1∶3)纯化得白色固体4 7.54 g,收率79%,m.p.150℃ ~152℃;1H NMR δ:7.55~7.59 (m,2H),7.26(d,J=3.6 Hz,1H),7.14~ 7.21(m,5H),6.74(d,J=3.6 Hz,1H),5.29 (t,J=9.6 Hz,1H),5.02(t,J=9.8 Hz,1H),4.94(t,J=9.6 Hz,1H),4.74(t,J=4.8 Hz,1H),4.57(d,J=9.6 Hz,1H),4.12(s,3H),3.75~3.79(m,1H),3.49~3.52(m,1H),3.40~3.44(m,1H),2.25(s,3H),1.99(s,3H),1.90(s,3H),1.70(s,3H)。
(4)5的合成
在反应瓶中依次加入干燥CH2Cl250 mL,4 7.50 g(13.14 mmol)和 Et3N 4.01 g(39.67 mmol),搅拌使其溶解;冰水浴冷却,缓慢滴加MsCl(2.27 g)的CH2Cl2(10 mL)溶液,滴毕,于室温反应1 h(TLC监测)。小心倾入300 mL冰水中,用CH2Cl2(3×100 mL)萃取,合并有机相,依次用1%盐酸(2×100 mL)和饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂:A=1∶4)纯化得白色固体5 5.23 g,收率61%,m.p.157℃ ~159℃;1H NMR δ: 7.55~7.59(m,2H),7.27(d,J=3.6 Hz,1H),7.15~7.21(m,5H),6.74(d,J=3.2 Hz,1H),5.36(t,J=9.4 Hz,1H),5.08(t,J=9.8 Hz,1H),5.01(t,J=9.6 Hz,1H),4.67(d,J=9.6 Hz,1H),4.25~4.33(m,2H),4.14~4.18(m,1H),4.12(s,2H),3.11(s,3H),2.25(s,3H),2.02(s,3H),1.92(s,3H),1.71(s,3H)。
(5)6的合成
在反应瓶中依次加入DMF 25 mL,5 5.13 g (7.91 mmol)和KI 6.70 g(40.35 mmol),搅拌使其溶解;于80℃反应12 h(TLC监测)。稍冷后小心倾入300 mL冰水中,用CH2Cl2(3×100 mL)萃取,合并有机相,用饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂:A=1∶5)纯化得白色固体6 4.28 g,收率79.5%,m.p.154℃ ~156℃;1H NMR δ: 7.55~7.59(m,2H),7.28(d,J=3.6 Hz,1H),7.16~7.21(m,5H),6.76(d,J=3.6 Hz,1H),5.35(t,J=9.4 Hz,1H),4.92(t,J=9.2 Hz,1H),4.90(t,J=9.6 Hz,1H),4.69(d,J= 10.0 Hz,1H),4.12(s,2H),3.69~3.73(m,H),3.49(dd,J=2.8 Hz,11.2 Hz,1H),3.24~3.26(m,1H),2.26(s,3H),2.02(s,3H),1.90(s,3H),1.71(s,3H)。
(6)7的合成
在反应瓶中依次加入干燥甲苯30 mL,6 4.20 g(6.17 mmol),AIBN 1.04 g(6.31 mmol)和n-Bu3SnH 5.51 g(18.94 mmol),氮气保护下于室温反应过夜(TLC监测)。小心倾入200 mL水中,用CH2Cl2(3×100 mL)萃取,合并有机相,依次用饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析 (洗脱剂:A= 1∶6)纯化得白色固体7 2.38 g,收率69.5%,m.p.149℃ ~151℃;1H NMR δ:7.55~7.59 (m,2H),7.27(d,J=3.6 Hz,1H),7.15~7.21(m,5H),6.74(d,J=3.6 Hz,1H),5.26 (t,J=9.6 Hz,1H),4.97(t,J=9.6 Hz,1H),4.81(t,J=9.6 Hz,1H),4.55(d,J=9.6 Hz,1H),4.12(s,2H),3.82~3.86(m,1H),2.24 (s,3H),2.02(s,3H),1.91(s,3H),1.70(s,3H),1.12(d,J=6.4 Hz,3H)。
(7)8的合成
在反应瓶中依次加入MeOH 50 mL和7 2.30 g(4.15 mmol),搅拌使其呈悬浮液;加入30% NaOH溶液10 mL,回流反应0.5 h(TLC监测)。稍冷后倾入300 mL冰水中,用浓盐酸调至pH 7~8,用乙酸乙酯(3×50 mL)萃取,合并有机相,用饱和食盐水(2×100 mL)洗涤,无水Na2SO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂:乙酸乙酯)纯化得白色固体8 1.53 g,收率86%,m.p. 139℃ ~141℃;1H NMR δ:7.56~7.60(m,2H),7.26(d,J=3.6 Hz,1H),7.17~7.21(m,3H),7.10~7.12(m,2H),6.78(d,J=3.6 Hz,1H),4.92(d,J=5.2 Hz,1H),4.86(d,J=4.0 Hz,1H),4.68(d,J=5.2 Hz,1H),4.14(d,J= 16.0 Hz,1H),4.09(d,J=16.0 Hz,1H),3.96 (d,J=9.2 Hz,1H),3.14~3.31(m,3H),2.90~2.95(m,1H),2.25(s,3H),1.15(d,J= 6.0 Hz,3H);13C NMR δ:162.54,160.12,143.57,140.19,138.23,137.37,134.85,130.47,129.64,128.85,126.94,126.86,126.31,126.05,123.34,115.93,115.72,81.32,78.16,75.71,75.59,74.81,33.35,18.75,18.23;ESI-MS m/z: 446.05{[M+NH4]+}。
2 结果与讨论
2.1 合成
(1)4的合成
4的合成由3在80%醋酸溶液中于40℃脱去TBDMS保护基而得。研究表明过高的反应温度会极大地增加反应速度,但是很大部分4会发生乙酰基从4-O-位转位到6-O-位。
(2)8的合成
在8的合成中,7在MeOH中经30%NaOH溶液水解脱去乙酰基。值得注意的是,由于水解后反应混合物中产生了醋酸盐,因此在用乙酸乙酯萃取前pH不可调得过低(<3),否则水解产生的醋酸盐会在酸性条件下被萃取到有机相中而在终产物8中残留超标。
2.2 1和8的生物活性
按文献[5]方法测定1和8体外对hSGLT2和hSGLT1抑制的IC50值,结果见表1。
由表1可见,1和8的IC50(hSGLT2)分别为3.9 nM和4.8 nM;IC50(hSGLT1)分别为695 nM和929 nM,对SGLT1的选择性(SGLT2/SGLT1)分别为178和194。可见1经6-OH脱氧成8后,对hSGLT2的抑制活性略有减弱,但变化不大。但是脱氧后对hSGLT1的抑制减弱了很多,因此导致脱氧产物8对hSGLT2的选择性有一定增加。综合对hSGLT2的抑制活性和hSGLT2/hSGLT1选择性的评价可以看出,8相对于母体化合物1具有相近的成药性,说明1中6-OH对维持其活性不是必要的,即6-OH不是关键药效团,这个结论与前述[5]6-OH不是Dapagliflozin的关键药效团的结论一致。

表1 1和8体外对hSGLT2和hSGLT1的抑制活性Table 1 In vitro inhibition activities of 1 and 8 against hSGLT1 and hSGLT2
按文献[5]方法测定了8对大鼠的尿糖UGE值,结果见表2。由表2可见,在大鼠尿糖试验中,8在用药量分别为2 mg·kg-1,6 mg·kg-1,18 mg·kg-1和54 mg·kg-1时,均能诱导出比母体化合物1更多的尿糖,但除6 mg·kg-1外(p<0.05),其余各剂量均无统计学意义上的显著性差异(p>0.05)。

表2 1和8的大鼠尿糖测定结果Table 2 The rat UGE results of 1 and 8
3 结论
生物活性测试结果显示,1的6-OH脱氧产物8比母体化合物1的体外活性稍弱,但体内活性稍强(这种体内外活性的微小差异可能与两者的药代性质的差异有关,8的药代性质应该略好于1),总体上认为两者的活性并无显著差异,说明1中葡萄糖片段中的6-OH不是关键药效团,这对于设计新的SGLT2抑制剂具有重要的指导意义。
[1]Washburn W N.Development of the renal glucose reabsorption inhibitors:A new mechanism for the pharmacotherapy of diabetes mellitus type 2[J].J Med Chem,2009,52:1785-1794.
[2]Hardman T C,Dubrey S W.Development and potential role of type-2 sodium-glucose transporter inhibitors for management of type 2 diabetes[J].Diabetes Ther,2011,2:133-145.
[3]Meng M,Ellsworth B A,Nirschl A A,et al.Discovery of dapagliflozin:A potent,selective renal sodiumdependent glucose cotransporter 2(SGLT2)inhibitor for the treatment of type 2 diabetes[J].J Med Chem,2008,51:1145-1149.
[4]Nomura S,Sakamaki S,Hongu M,et al.Discovery of canagliflozin,a novel C-glucoside with thiophene ring,as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus[J].J Med Chem,2010,53:6355-6360.
[5]Zhang L,Wang Y,Xu H,et al.Discovery of 6-deoxydapagliflozin as a highly potent sodium-dependent glucose cotransporter 2(SGLT2)inhibitor for the treatment of type 2 diabetes[J].Med Chem,in press.
[6]Ch R,Tyagi M,Patil P R,et al.DABCO:An efficient promoter for the acetylation of carbohydrates and other substances under solvent-free conditions[J]. Tetrahedron Lett,2011,52:5841-5846.
6-羟基脱氧Canagliflozin的合成及其生物活性*
韩书文1,2,徐华强2,3,王玉丽2,张玲钰2,3,史永恒2,刘冰妮2,魏群超2,徐为人1,2,赵桂龙2
Synthesis and Biological Activities of 6-Deoxylated Canagliflozin
HAN Shu-wen1,2, XU Hua-qiang2,3, WANG Yu-li2, ZHANG Ling-yu2,3,SHI Yong-heng2, LIU Bing-ni2, WEI Qun-chao2, XU Wei-ren1,2, ZHAO Gui-long2
(1.Graduate School,Tianjin University of Traditional Chinese Medicine,Tianjin 300193,China;2.Tianjin Key Laboratory of Molecular Design and Drug Discovery,Tianjin Institute of Pharmaceutical Research,Tianjin 300193,China;3.School of Chemistry and Chemical Engineering,Shandong University,Jinan 250100,China)
6-Deoxylated Canagliflozin,4-(6-deoxy-β-D-glucopyranosyl)-2-[5-(4-fluorophenyl)thiophen-2-yl]-1-toluene(8),a novel SGLT2 inhibitor,was synthesized by a seven-step reaction from Canagliflozin(1)in overall yield of 21%.The structure was characterized by1H NMR,13C NMR and MS.In vitro assays showed that the properties of 1 and 8 were as follows:IC50(hSGLT2)were 3.9 nM and 4.8 nM,selectivity(SGLT2/SGLT1)were 178 and 194,respectively.In vivo primary biological assays exhibited that 8 could induce a considerable amount of urinary glucose in rat urinary glucose excretion test,however it was not statistically different from 1.
SGLT2 inhibitor;Canagliflozin;deoxy;synthesis;inhibition activity;urinary glucose excretion text(UGE)
O629.11;O626.1;O625.1
A
1005-1511(2014)01-0001-05
2013-05-02;
2013-12-13
国家自然科学基金资助项目(21302141);天津市科技支撑计划重点项目(10ZCKFSH01300)
韩书文(1986-),男,汉族,黑龙江齐齐哈尔人,硕士研究生,主要从事药物化学的研究。
赵桂龙,副研究员,Tel.022-23006869,E-mail:zhao_guilong@126.com
