火焰原子吸收分光光度法测定五氧化二钒中铁及氧化钾、氧化钠含量
2014-07-24韩晋园马增敏刘亚莉刘志虹胡锦川孙明力毕京红
韩晋园,马增敏,刘亚莉,刘志虹,胡锦川,孙明力,毕京红
(1.北京北方车辆集团有限公司计量理化中心 北京 100072; 2.中国人民解放军驻六一八厂军代室,北京 100072)
火焰原子吸收分光光度法测定五氧化二钒中铁及氧化钾、氧化钠含量
韩晋园1,马增敏1,刘亚莉1,刘志虹1,胡锦川2,孙明力1,毕京红1
(1.北京北方车辆集团有限公司计量理化中心 北京 100072; 2.中国人民解放军驻六一八厂军代室,北京 100072)
五氧化二钒样品用盐酸分解,在稀盐酸介质中,用原子吸收分光光度计分别于248.3,766.5,589.0 nm波长处,使用空气-乙炔火焰,测量五氧化二钒中铁及氧化钾、氧化钠含量。在最佳实验条件下,铁、氧化钾、氧化钠的质量浓度分别在0.05~0.20,0.05~0.80,0.20~1.0 mg/L范围内与吸光度线性关系良好,相关系数分别为0.998 6,0.994 3,0.994 2。方法检出限铁为6.7 μg/L,氧化钾为1.0μg/L,氧化钠为1.4 μg/L,加标回收率为95.9%~103.0%。铁、氧化钾、氧化钠测定结果的相对标准偏差分别为3.2%,4.2%,2.9% (n=6)。该方法适合五氧化二钒中铁及氧化钾、氧化钠的测定。
火焰原子吸收分光光度法;五氧化二钒;铁;氧化钾;氧化钠
金属钒素有金属“维生素”之称,具有众多优异的物理性能和化学性能,被广泛用于黑色、有色合金和催化剂等领域。自然界中钒很难以单一体存在,主要与其它矿物形成共生矿或复合矿,主要的矿物有钒钛磁铁矿、钾钒铀矿、石油伴生矿。我国钒储量十分丰富,主要为钒钛磁铁矿和石煤。就我国目前提炼钒的技术而言,一般含钒量大于0.8%的石煤才具有开发利用价值[1]。在YB/T 5304-2006 《五氧化二钒》中,五氧化二钒按用途和品位主要分为99,98,97三个牌号及各牌号对应的主要杂质元素含量,同时根据特殊要求,可提供杂质含量更低产品及其它杂质元素的实测数据[2]。传统方法用邻二氮杂菲分光光度法测定五氧化二钒中铁量[3],用原子吸收分光光度法测定五氧化二钒中氧化钾、氧化钠含量[4]。这些方法中有些操作步骤烦琐费时,难以满足现代企业生产的要求。原子吸收技术的发展为同时测定多种元素提供了方便。目前分析铁的方法很多,如原子吸收法(AAS)、邻菲罗啉分光光度法、电感耦合等离子体发射光谱法(ICP—AES),常量铁还可以用高锰酸钾滴定法分析[5-10]。笔者用火焰原子吸收分光光度法连续测定五氧化二钒中铁、氧化钾、氧化钠,该法方便快捷,测定结果可靠,适于大批量样品的测定。
1 实验部分
1.1 主要仪器与试剂
原子吸收分光光度计:WFX-120型,北京瑞利分析仪器公司;
铁、钾、钠空心阴极灯:北京有色金属研究总院;
铁标准溶液:10 μg/mL,称取0.100 0 g铁丝(纯度大于99.99%)于500 mL烧杯中,加入5 mL王水,加热溶解,冷却后加入10 mL硫酸溶液(1+4),加热蒸发至冒三氧化硫白烟,冷却。加入50 mL水,加热使盐类溶解,冷却至室温,移入1 000 mL容量瓶中,以水稀释至标线,混匀。移取10.00 mL上述铁标准溶液,置于100 mL容量瓶内,用水稀释至标线,混匀;
氧化钾标准溶液:10 μg/mL,称取0.158 3 g预先经500℃灼烧0.5 h并置于干燥器中冷却至室温的基准氯化钾,溶于水中,加入10 mL盐酸溶液(1+4),移入1 000 mL容量瓶内,用水稀释至标线,混匀。移入干塑料瓶内储存备用(此溶液质量浓度为100 μg/mL)。移取10.00 mL上述氧化钾标准溶液置于100 mL 容量瓶内,用水稀释至标线,混匀;
氧化钠标准溶液:10 μg/mL,称取0.188 6 g预先经500℃灼烧0.5 h并置于干燥器中冷却至室温的基准氯化钠,溶于水中,加入10 mL盐酸溶液(1+4),移入1 000 mL容量瓶内,用水稀释至标线,混匀。移入干塑料瓶内储存备用(此溶液质量浓度为100 μg/mL)。移取10.00 mL上述氧化钠标准溶液置于100 mL 容量瓶内,用水稀释至标线,混匀;
五氧化二钒溶液:10 mg/mL,称取1.000 g高纯五氧化二钒,溶于40 mL盐酸溶液(1+1)中,移入100 mL容量瓶中,用水稀释至标线,混匀;
实验所用试剂均为分析纯,水为实验室用二级水。
1.2 仪器工作条件
仪器最佳工作条件见表1。
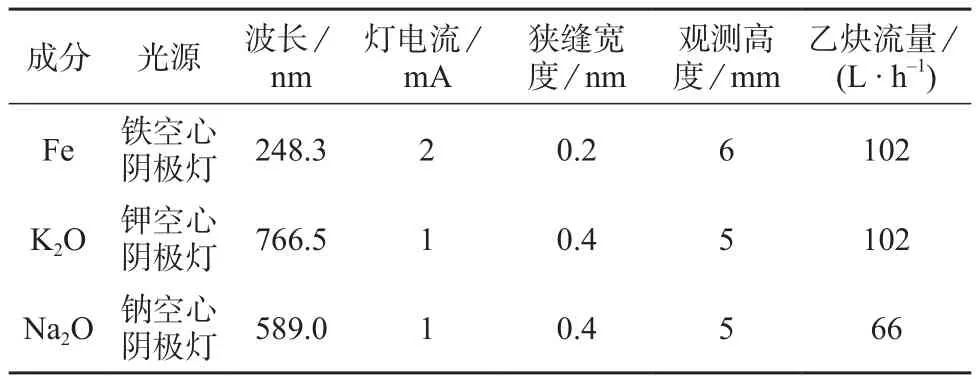
表1 仪器工作条件
1.3 样品
选取5个实验室所测的五氧化二钒样品,其主要化学成分见表2
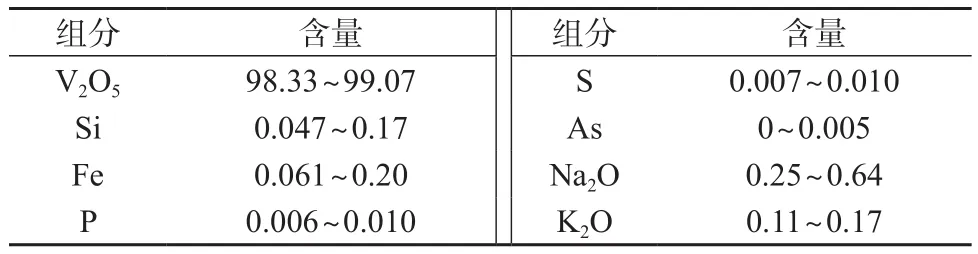
表2 五氧化二钒主要成分含量参考值 %
1.4 样品处理
称取0.1 g试样(精确至0.000 1 g)置于100 mL烧杯中,加入20 mL盐酸溶液(1+4),加热使其溶解,冷却后移入100 mL容量瓶中,用水稀释至标线,混匀,得样品溶液。随同作空白溶液。
1.5 样品测定
将1.4样品溶液稀释(测铁时用原液,测氧化钾、氧化钠时稀释5倍),按1.2仪器工作条件,以水调零,测量样品溶液及空白溶液中铁、氧化钾、氧化钠的吸光度。从工作曲线上查得相应的铁、氧化钾、氧化钠的质量浓度。
2 结果与讨论
2.1 溶样酸的选择
分别采用不同配比的盐酸溶液对标准物质GSB 03-1688-2004(标准值为Fe 0.16%,Na2O 1.11%,K2O 0.15%)进行溶解,用火焰原子吸收法测定样品溶液中铁、氧化钾、氧化钠含量,结果见表3。
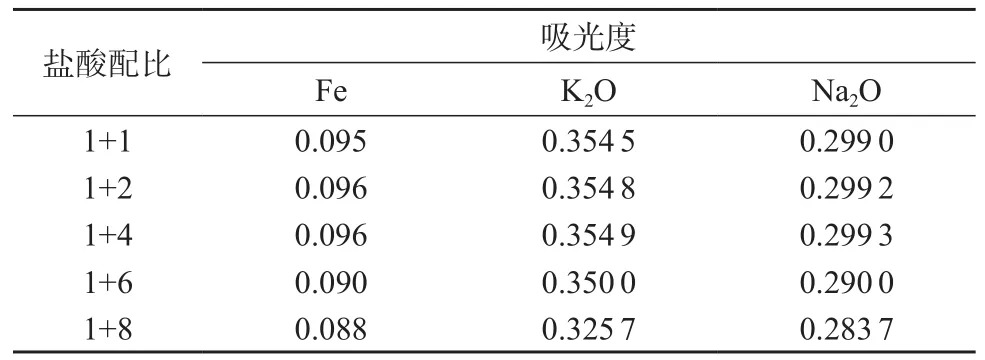
表3 盐酸溶液配比不同时的吸光度对比
由表3可知,盐酸溶液(1+2)与(1+4)吸光度相差不大,但盐酸用量少,可减少对燃烧头的腐蚀,而更稀的盐酸溶液溶样速度慢,所以实验选用盐酸溶液(1+4)溶样。
2.2 工作曲线
于一组100 mL容量瓶中,配制系列混合标准溶液如表4所示。溶液中均加入2 mL盐酸溶液(1+4),补加5.0 mL五氧化二钒溶液。按1.2仪器条件测定系列混合标准溶液中铁及氧化钾、氧化钠的吸光度,以铁及氧化钾、氧化钠的浓度c为横坐标,吸光度A为纵坐标绘制工作曲线,回归方程和相关系数见表5。以平行测定11次试剂空白结果的3倍标准偏差计算方法检出限,结果见表5。
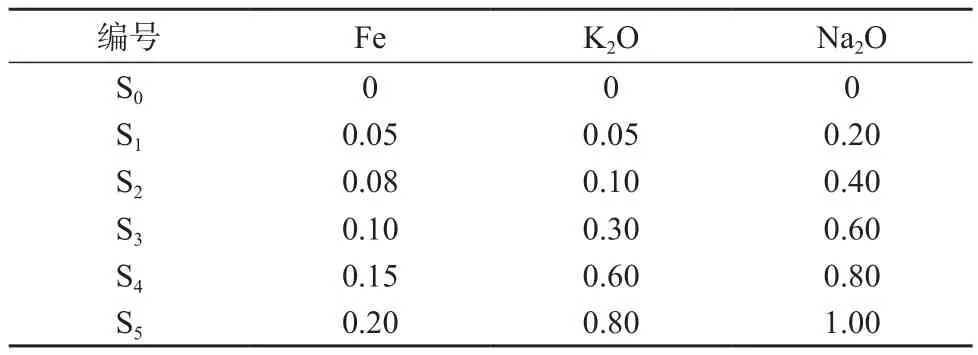
表4 系列混合标准溶液 mg/L
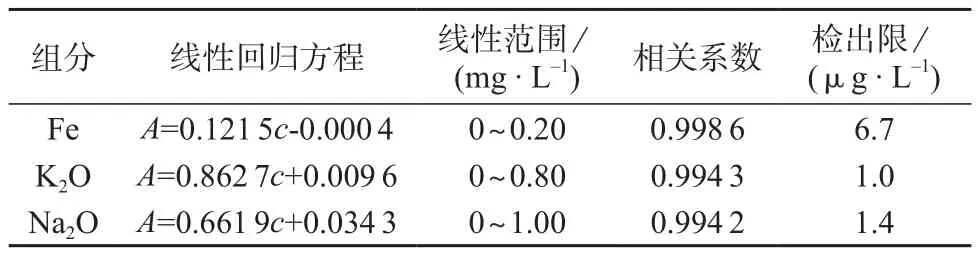
表5 线性回归方程和方法检出限
2.3 干扰试验
2.3.1 五氧化二钒基体的影响
根据各元素含量的不同,在一组100 mL容量瓶中,分别在含0.80 mg/L铁、0.4 0 mg/L氧化钾、0.40 mg/L氧化钠的混合标准溶液中加入不同量的五氧化二钒溶液,进行干扰试验,结果见表6。由表6可知,样品中50.0%~90.0%的五氧化二钒均不干扰铁、氧化钾、氧化钠的测定。
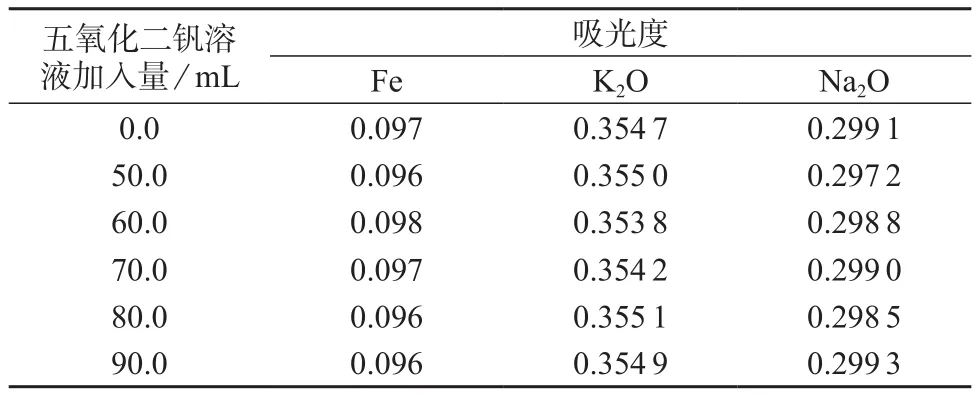
表6 五氧化二钒基体的干扰试验结果
2.3.2 其它共存离子的影响
根据各元素在自然界可能存在含量的不同,在一组100 mL容量瓶中,分别在含0.08 mg/L铁、0.40 mg/L氧化钾、0.40 mg/L氧化钠的混合标准溶液中加入一定量的Ca,Pb,Mn等共存元素进行干扰试验。结果表明:100 mL标准溶液中,低于20 mg Ca,5 mg Pb,3 mg Mn均不干扰铁及氧化钾、氧化钠的测定。实际遇到的五氧化二钒中以上元素含量均不会高于上述值。
2.4 加标回收试验
在处理后的试液中加入一定量的铁及氧化钾、氧化钠标准溶液进行回收试验,结果见表7。由表7可知,铁及氧化钾、氧化钠的加标回收率为95.9%~103.0%,说明该方法具有较高的准确度。
2.5 精密度试验
选择五氧化二钒标准样品(GSB03-1688-2004)按分光光度法测定铁量进行比较,结果见表8。由表8可知,本法测定结果的相对标准偏差为2.9%~3.2%,表明本法精密度较好。

表7 回收试验结果
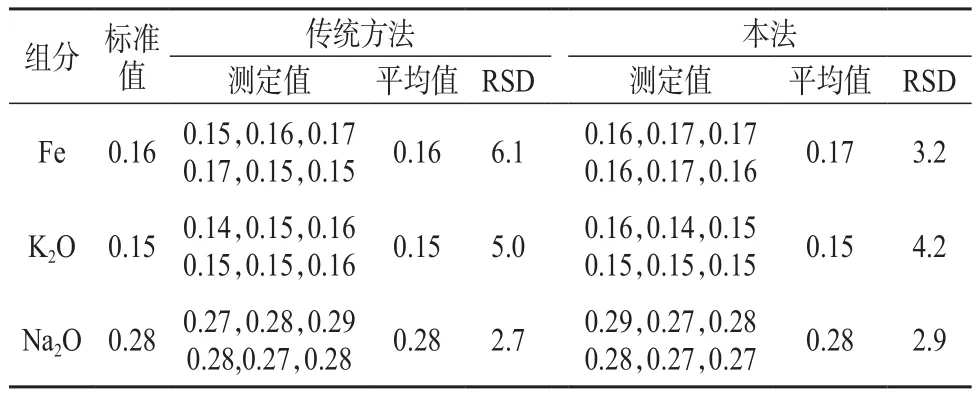
表8 精密度试验结果 %
3 结语
五氧化二钒样品用盐酸分解后,用火焰原子吸收光谱法测定其中的铁及氧化钾、氧化钠的含量。该法具有操作简便、检出限低、精密度和准确度高的特点,能够满足五氧化二钒的检验要求。与传统方法邻二氮杂菲分光光度法测定铁量进行比较相比,本法精密度较好。
[1]冯其明,何东升.国内外金属矿山低品位矿选矿新技术进展——以铝、铜、镍、钒为例[J].金属矿山,2008(S1): 51-62.
[2]YB/T 5304-2006 氧化二钒[S].
[3]YB/T 5330-2006 五氧化二钒化学分析方法邻二氮杂菲分光光度法测定铁量[S].
[4]YB/T 5335-2006 五氧化二钒化学分析方法原子吸收分光光度法测定氧化钾和氧化钠量[S].
[5]刘琳娟,张琪,陆培培.标准加入-原子吸收光谱法测定钢渣中的铁[J].岩矿测试,2010,29(6): 695-698.
[6]洪建和,安黛宗,田曙玲.高锰酸钾滴定法测定常量铁的改进[J].实验室研究与探索,2008,16(8): 12-14.
[7]汾宗平.电感藕合等离子体发射光谱法测定氧化钒中钾钠硫磷铁[J].冶金分析,2010,30(3): 30-33.
[8]唐杰,魏成富,杨梨容,等.原子吸收光谱法测定TC11 合金中微量铁[J].绵阳师范学院学报,2010,29(11): 35-37.
[9]林庆光.原子吸收分光光度法与二氮杂菲分光光度法测水中铁的比较[J].中国热带医学,2008,8(6): 1 034-1 052.
[10]王玉宝.火焰原子吸收光谱法测定含硅铝合金中微量铬铜镁铁[J].冶金分析,2007,27(10): 50-52.
Determination of Iron,Sodium Oxide and Potassium Oxide Content in Vanadium Pentoxide by Flame Atomic Absorption Spectrometry
Han Jinyuan1, Ma Zengmin1, Liu Yali1, Liu Zhihong1, Hu Jinchuan2, Sun Mingli1, Bi Jinghong1
(1. Metrological Physicochemical Center, Beijing North Vehicle, Beijing 100072, China; 2. Chinese People’s Liberation Army Representative Office in 618 Factory, Beijing 100072, China)
The vanadium pentoxide sample was decomposed with hydrochloric acid. Then in diluted hydrochloric acid medium,the content of iron,sodium oxide and potassium oxide in vanadium pentoxide was determined by flame atomic absorption spectrometry at 248.3,766.5,589.0 nm,respectively. Under optimized experimental conditions,the absorbance was linear with the concentration of iron,sodium oxide and potassium oxide in the range of 0.05-0.20,0.05-0.80,0.20-1.0 mg/L,respectively. The correlation coefficients were 0.998 6,0.994 3,0.994 2,respectively. The detection limits for iron ,sodium oxide and potassium oxide were 6.7,1.0,1.4 μg/L,respectively. The recoveries ranged from 95.9% to 103.0%. The relative standard deviations of iron,sodium oxide and potassium oxide determination results were 3.2%,4.2%,2.9%(n=6), respectively. This method was applicable for the determination of iron,sodium oxide and potassium oxide content in vanadium pentoxide.
flame atomic absorption spectrometry; vanadium pentoxide; iron; sodium oxide; potassium oxide
O657.3
A
1008-6145(2014)04-0067-03
10.3969/j.issn.1008-6145.2014.04.020
联系人:韩晋园;E-mail: hjy3721@sohu.com
2014-04-23
