硫化氢吸入干预大鼠棉花烟雾吸入性肺损伤中的氧化应激
2014-07-18韩志海王晓阳方庭正段蕴铀
姜 毅,韩志海,王晓阳,方庭正,黄 燕,段蕴铀
硫化氢吸入干预大鼠棉花烟雾吸入性肺损伤中的氧化应激
姜 毅1,2,韩志海3,王晓阳1,方庭正1,黄 燕1,段蕴铀1
目的探讨吸入硫化氢(hydrogen sulfide, H2S)干预大鼠棉花烟雾吸入性肺损伤的氧化应激反应机制。方法24只雄性SD大鼠随机分成对照组、H2S组、烟雾组、烟雾+H2S组,每组6只。复制大鼠棉花烟雾吸入性损伤模型,在烟雾吸入或模拟烟雾吸入后,H2S组、烟雾+H2S组大鼠予以持续吸入H2S 80 ppm+30%氧气6 h,对照组、烟雾组予以吸入30%氧气6 h,ELISA法检测肺组织匀浆中MDA、NO、iNOS、NF-κB p65浓度,免疫组化检测肺组织NF-κB p65并进行半定量分析,荧光定量PCR法行肺组织iNOS mRNA定量。结果烟雾组大鼠肺组织匀浆中MDA、NO、iNOS、NF-κB p65浓度和肺组织中NF-κB p65的累积光密度、iNOS mRNA的相对表达量均明显高于对照组,而烟雾+H2S组的上述指标较烟雾组均明显降低,如肺组织匀浆中NF-κB p65浓度(8123.51±2095.33) pg/mlvs(13803.19±2196.37) pg/ml,P<0.001;肺组织中iNOS mRNA的相对表达量(1.04±0.24)vs(2.20±0.21),差异有统计学意义(P<0.001);H2S组iNOS浓度、iNOS mRNA的相对表达量、NF-κB p65的累积光密度高于对照组,但MDA、NO、iNOS、NF-κB p65浓度与对照组比较无明显差别。结论吸入H2S的干预机制可能是吸入H2S可抑制NF-κB p65的激活,使iNOS mRNA的转录合成减少,从而减少iNOS、NO生成,减轻氧化应激反应和减轻大鼠肺损伤。
急性肺损伤;烟雾吸入性损伤;氧化应激;硫化氢
烟雾吸入性损伤主要发生在呼吸道和肺实质,严重者产生中毒性肺炎或肺水肿,可迅速进展为急性肺损伤/急性呼吸窘迫综合征(acute lung injury/acute respiratory distress syndrome,ALI/ARDS),使救治难度加大,病死率增加[1]。在烟雾吸入性损伤的发病机制中,氧化应激占有很大比重。高温烟雾中的很多成分是强氧化剂,并且炎性反应失控时大量的炎性反应细胞聚集在肺内,产生过量的活性氧(reactive oxygen species,ROS),也可诱导氧化应激损伤。
近十年来的研究发现,H2S具有抗氧化应激[2]、调节炎性反应[3]、舒张血管[4-7]、抗纤维化、参与调节内分泌及生殖系统功能等作用[8]。吸入H2S 80 ppm 6 h,在内毒素诱导的ALI小鼠模型中可抑制全身炎性反应,提高小鼠生存率[9, 10];在过度通气诱导的ALI小鼠模型中可抑制肺内炎性反应和肺泡上皮细胞凋亡而保护肺脏[11]。本课题前期实验研究证实,大鼠棉花烟雾吸入性肺损伤时氧化应激反应增强,本实验经预实验后观察吸入80 ppm H2S 6 h气体对大鼠烟雾吸入性肺损伤时氧化应激反应的影响。
1 材料与方法
1.1 实验动物及分组 清洁级健康成年雄性SD大鼠共24只,体重150~250 g,由军事医学科学院实验动物中心[SCXK-(军)-2012-0004]提供,海军总医院实验动物中心[SCXK-(军)-2012-0012]饲养。遵守实验动物条例处置动物,按照随机化原则将动物分组,分为对照组、H2S组、烟雾组、烟雾+H2S组,每组6只。
1.2 大鼠烟雾吸入性损伤模型的复制及H2S的吸入 参照文献[12-14],本课题组已成功制作大鼠烟雾吸入性损伤模型,操作如下:大鼠2只分别置入吸烟瓶中,锡炉预热至300 ℃恒温,称取2 g棉花放入锡炉内,立即盖上集烟筒,启动风扇,开始计时2 min,见大鼠足底皮肤逐渐出现樱红色至紫红色,并有躁动、呼吸急促、张口呼吸,至出现呼吸深慢、张口喘鸣时或满2 min时限时,予以停止吸烟,立即敞开吸烟瓶口,吸入空气7 min,重复上述步骤3~5次,至大鼠吸入空气7 min仍旧昏迷不醒时结束。4组大鼠均先后置于烟雾吸入装置中给予上述类似处置,其中烟雾组、烟雾+H2S组给予烟雾输入,对照组、H2S组无烟雾输入;在烟雾吸入或模拟烟雾吸入后,4组大鼠均给予30%氧气吸入6 h,但H2S组、烟雾+H2S组大鼠还同时予以吸入H2S 80 ppm 6 h。实验中大鼠予以自由进食及饮水。
1.3 ELISA检测 实验结束后大鼠腹腔注射戊巴比妥钠致死。采用双抗体夹心ABC-ELISA法检测右肺下叶匀浆中一氧化氮(nitric oxide,NO)、诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、核转录因子-κB p65(nuclear factor-kappaB,NF-κB p65)浓度。比色法检测右肺下叶肺组织匀浆中丙二醛(malondialdehyde,MDA)浓度。ELISA试剂盒厂家为嘉美生物(Jiamay Biotech Co.Ltd),按试剂盒说明操作。
1.4 右肺中叶肺组织NF-κB p65免疫组化检测及半定量分析 右肺中叶4%多聚甲醛溶液浸泡72 h后常规石蜡包埋、切片,厚度3 μm,60~65 ℃烤片4 h,脱蜡、水化、PBS缓冲液洗涤,高压修复组织抗原,3%H2O2灭活过氧化物酶,正常山羊血清封闭,滴加第一抗体Anti-NF-κB p65 antibody (abcam,ab16502) 50 μl,稀释度1∶200,4 ℃孵育过夜,滴加复合二抗HRP-Polymer anti Mouse/Rabbit IgG(Maixin.Bio,KIT-5020),室温静置20 min;DAB显色,苏木精复染,阴性对照以血清代替一抗。镜检阳性染色为黄色或棕黄色染色。应用Image Pro Plus 6.0图像分析系统(美国Media Cybernetics公司)进行半定量分析,每张玻片显微镜下随机选择5个高倍镜视野(×1000),以相同参数摄取图像,IPP6.0软件分析得出每个视野阳性染色的平均光密度(mean density)、积分光密度(integrated optical density,IOD)的总和即累积光密度(sum IOD),计算出每个标本上述指标的均值,从而对染色浓度进行半定量分析。
1.5 荧光定量PCR方法检测大鼠右肺上叶iNOS mRNA转录水平 目的基因iNOS的PCR检测引物序列由嘉美生物设计合成,iNOS上下游引物分别是5′-ACACCGATTCCACTCAACTA-3′和5′-ACCACCTGTTAGTTCAAGCC-3′,扩增产物长度为159bp,内参为β-actin(CWbio.Co.Ltd,Cat#CW0918)。用超纯RNA提取试剂盒(CWbio.Co.Ltd,Cat#CW0581)提取组织样本中总RNA。取5 μl RNA用1%琼脂糖凝胶进行电泳。用HiFi-MMLVcDNA第1链合成试剂盒(CWbio.Co.Ltd,Cat#CW0744)进行反转录,用UltraSYBR Mixture(with Rox)(CWbio.Co.Ltd,Cat#CW0956)进行扩增,扩增程序为:95 ℃ 10 min,(95 ℃15 s+60 ℃ 60 s) 40个循环。用LightCycler-480II型荧光定量PCR仪测量,采用2-△△CT法进行数据的相对定量分析。
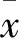
2 结 果
2.1 大鼠肺组织匀浆ELISA检测结果 由表1可见,烟雾组大鼠肺组织匀浆中MDA、NO、iNOS、NF-κB p65浓度较对照组均明显升高(P<0.001),烟雾+H2S组上述指标较烟雾组降低(P<0.001)。H2S组上述指标仅iNOS浓度高于对照组,其他指标与对照组比较无明显差别,H2S组上述指标均低于烟雾组。
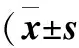
表1 各组大鼠肺组织中MDA、NO、iNOS、NF-κB p65浓度检测结果 ;n=6)
注:与烟雾组比较,①P<0.05;与对照组比较, ②P<0.05
2.2 大鼠体重和肺组织iNOSmRNA的相对表达量 由表2可见,各组大鼠体重没有统计学差异(P>0.05),大鼠平均体重为(186.68±28.79) g,可以认为大鼠体重对各组间实验结果没有影响。烟雾组、烟雾+H2S组和H2S组大鼠肺组织iNOSmRNA的相对表达量较对照组明显升高(P<0.001),烟雾+H2S组和H2S组较烟雾组降低(P<0.001)。
2.3 大鼠肺组织iNOS的免疫组化分析结果 大鼠肺组织NF-κB p65的免疫组化染色结果见图1。由表2可见,大鼠肺组织NF-κB p65的累积光密度(sum IOD)烟雾组较对照组高(P<0.001);烟雾+H2S组和H2S组较烟雾组低(P<0.01),但比对照组高(P<0.001)。大鼠肺组织NF-κB p65的平均光密度(mean density) H2S组、烟雾组、烟雾+H2S组无统计学差异(P>0.05),但均低于对照组(P<0.01)。
表2 各组大鼠体重、肺组织iNOS mRNA相对表达量、NF-κB p65累积吸光度和平均光密度的比较 ;n=6)
注:与烟雾组比较,①P<0.05;与对照组比较, ②P<0.05
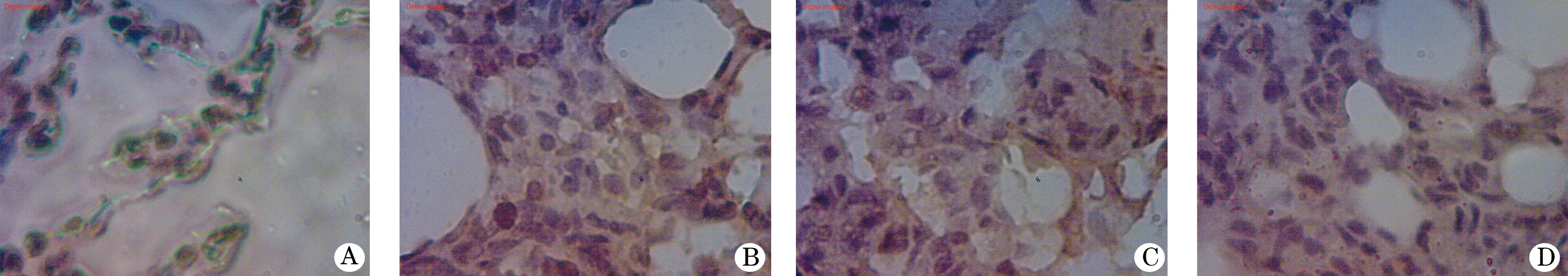
图1 肺组织NF-κB p65免疫组化染色镜下观察(×1000)
3 讨 论
烟雾成分的复杂性决定了烟雾吸入性肺损伤的发病机制的复杂性,尤以氧化应激更为重要[15]。烟雾吸入可激活肺内巨噬细胞、中性粒细胞、内皮细胞及血管平滑肌细胞释放出大量细胞因子,如肿瘤坏死因子-α(TNF-α)、白介素-1β(IL-1β)、IL-6、IL-8等,从而激活NF-κB,其在胞浆中分解后的活性片段NF-κB p65进入细胞核,促进下游iNOS mRNA的转录,导致iNOS的合成增加,iNOS可分解精氨酸产生大量NO,可催化合成过氧化亚硝酸盐,引起生物膜的脂质过氧化反应[16]。同时,烟雾中的NO、N2O、SO2、氧化性颗粒物均是强氧化剂,粒细胞受刺激30 s内即可释放出大量的氧自由基,缺氧后吸氧治疗也增加氧自由基的产生。氧自由基同样可使生物膜发生脂质过氧化反应,破坏膜结构,激活炎性介质的合成,影响能量代谢,并使蛋白质变性而发生功能障碍。上述损伤性因素使肺泡-毛细血管通透性增高,血液中有形成分渗出到肺泡腔,形成肺水肿。此外, 烟雾吸入性肺损伤产生的活性氧可导致NO合成过量[17],NO可使血管外漏、缺氧性肺血管收缩功能丧失[18],同时使具有细胞毒性的活性氮(reactive nitrogen species,RNS)生成增加[19],进一步加重肺损伤。既往本课题组的实验研究已证实,大鼠棉花烟雾吸入后6 h即可出现典型的肺损伤表现,本实验发现,烟雾吸入后6 h大鼠肺组织中NF-κB p65浓度、NF-κB p65的累积光密度、iNOSmRNA的相对表达量、iNOS和NO浓度均增高,完全符合上述烟雾吸入后氧化应激反应的表现,即烟雾吸入后激活NF-κB,NF-κB p65浓度增加并进入细胞核,促进iNOS mRNA的转录增加,使iNOS表达增加和NO合成增多。同时,MDA作为自由基与生物膜多价不饱和脂肪酸发生反应而生成的过氧化物,其含量的同步升高也说明氧化应激反应加剧,脂质过氧化反应增强,组织损伤加重。
火灾后烟雾吸入性肺损伤的早期救治尤为关键,从其发病机制可以看出,打断氧化应激反应通路,抑制NF-κB的激活从而减少其下游产物的生成,从理论上看有可能减轻肺损伤。有研究表明,连续静脉点滴低剂量精氨酸血管加压素,抑制iNOS产生过量的NO,可明显减轻烧伤和烟雾吸入导致的肺损伤[20]。
近些年的研究中,H2S一改既往“具有臭鸡蛋气味的神经毒气”形象,被称为继NO、CO之后的第3种气体信号分子[21, 22]。动物实验证实,静脉点滴硫氢化钠(NaHS)或吸入H2S气体,在多种原因所致的肺损伤动物模型研究中具有抗氧化应激、抗炎性反应、抗凋亡、减轻肺损伤的作用[2, 9-11, 23],但吸入H2S气体对烟雾吸入性肺损伤的作用研究尚无报道。在脂多糖激活的离体巨噬细胞中,H2S可抑制NF-κB信号通路的激活,减少NO的产生,而起到抗氧化应激的作用[24]。因此,笔者设计实验研究吸入H2S气体对烟雾吸入性肺损伤的干预效果。
实验发现,在烟雾吸入后立即予以吸入H2S 80 ppm 6 h,可明显减轻大鼠的肺损伤,在大鼠肺组织中NF-κB p65浓度、NF-κB p65的累积光密度、iNOSmRNA的相对表达量、iNOS和NO浓度均明显降低,MDA下降,提示吸入H2S减轻大鼠棉花烟雾吸入性肺损伤的机制可能是吸入H2S抑制了NF-κB p65的激活,使iNOSmRNA的转录合成减少,减少了iNOS、NO的生成,从而减轻氧化应激反应,减轻了大鼠的肺损伤。大鼠肺组织NF-κB p65的平均光密度(mean density) 代表了切片上免疫组化阳性染色的深浅,但不能代表阳性染色的总量变化,因而无指示意义。
细胞类型、氧自由基的类型、NF-κB通路上众多氧化应激敏感位点的状态、上游的信号通路等因素的变化均可影响到NF-κB的激活,从而干预氧化应激反应的进程[25],因此,NF-κB是细胞对氧化应激状态极为敏感的核转录因子。H2S调节NF-κB蛋白活性的上游通路包括ERK通路[26,27]、p38-MAPK通路[28]、血红蛋白加氧酶-1和热休克蛋白70[24]等。近年来对H2S的作用机制的研究发现,H2S信号转导的蛋白质巯基化机制,即H2S通过与靶蛋白的半胱氨酸活性残基结合,发生蛋白质巯基化反应,改变蛋白质的活性,可能是H2S作用机制的中心环节[29,30]。在本实验的烟雾+H2S组,烟雾为外部刺激,吸入后作用在肺内巨噬细胞、中性粒细胞、内皮细胞及血管平滑肌细胞,释放出大量细胞因子,如TNF-α、IL-1β、IL-6、IL-8等。同时,烟雾中的氧化剂也导致细胞内氧自由基明显增加,上述因素可激活NF-κB通路,使iNOS的表达及NO浓度均显著升高,并使细胞处于强氧化状态。由于机体抗氧化调节不足以对抗氧化应激[31],机体损伤加剧。在此基础上,外源性H2S的吸入表现出抑制NF-κB通路的作用,使iNOS表达及NO合成减少,而减轻了大鼠的肺损伤。说明大鼠棉花烟雾吸入后立即吸入H2S 80 ppm 6 h可减轻大鼠的肺损伤,H2S的应用具有研究意义。
本实验中H2S组MDA、NO、iNOS、NF-κB p65浓度与对照组比较无统计学差异,大鼠未见明显的肺损伤,说明单纯吸入80 ppm H2S 6 h对健康大鼠是相对安全的。本实验充实了H2S吸入研究的应用范畴。H2S属较强的还原剂,气道吸入后可直接与氧化剂发生中和反应,它至少能与超氧阴离子、过氧化氢、超氧化氮及次氯酸4种氧自由基起反应[32, 33],从而保护膜结构免受自由基损伤,这也是吸入H2S气体治疗烟雾吸入性肺损伤的便利条件,在以后的研究中可继续探讨。在本次实验研究中,只设计了6 h的治疗时间段的比较,缺乏长期动态效果的观察。另外,H2S可激活ATP敏感钾通道而舒张血管[6],调节Fas/FasL死亡受体通路而减轻凋亡[34],抑制神经源性炎性反应,3种气体信号分子相互作用等[35],这些机制也可能在烟雾性肺损伤的治疗中发挥作用,可进一步探讨。
本研究揭示了吸入H2S 80 ppm 6 h减轻大鼠棉花烟雾吸入性肺损伤的机制可能是吸入H2S抑制了NF-κB p65的激活,使iNOSmRNA的转录合成减少,减少了NO的生成,减轻氧化应激反应,从而减轻了大鼠的肺损伤。
[1] Ballard-Croft C, Sumpter L R, Broaddus R,etal. Ovine smoke/burn ARDS model: a new ventilator-controlled smoke delivery system [J]. J Surg Res, 2010, 164(1): 155-162.
[2] Esechie A, Kiss L, Olah G,etal. Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation [J]. Clin Sci (Lond), 2008, 115(3): 91-97.
[3] Li P C, Chen W C, Chang L C,etal. Substance P(SP) acts via the neurokinin receptor 1 to elicit bronchoconstriction, oxidative stress, and upregulated ICAM-1 expression after oil smoke exposure [J]. Am J Physiol Lung Cell Mol Physiol, 2008, 294(5): 912-920.
[4] Papapetropoulos A, Pyriochou A, Altaany Z,etal. Hydrogen sulfide is an endogenous stimulator of angiogenesis [J]. Proc Natl Acad Sci USA, 2009, 106(51): 21972-21977.
[5] Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications [J]. Br J Pharmacol, 2011, 164(3): 853-865.
[6] Yang G, Wu L, Jiang B,etal. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase [J]. Science, 2008, 322(5901): 587-590.
[7] Muzaffar S, Jeremy J Y, Sparatore A,etal. H2S-donating sildenafil (ACS6) inhibits superoxide formation and gp91phox expression in arterial endothelial cells: role of protein kinases A and G [J]. Br J Pharmacol, 2008, 155(7): 984-994.
[8] Zhu X Y, Gu H, Ni X. Hydrogen sulfide in the endocrine and reproductive systems [J]. Expert Rev Clin Pharmacol, 2011, 4(1): 75-82.
[9] Tokuda K, Kida K, Marutani E,etal. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice [J]. Antioxid Redox Signal, 2012, 17(1): 11-21.
[10] Faller S, Zimmermann K K, Strosing K M,etal. Inhaled hydrogen sulfide protects against lipopolysaccharide-induced acute lung injury in mice [J]. Med Gas Res, 2012, 2(1): 26-31.
[11] Faller S, Ryter S W, Choi A M,etal. Inhaled hydrogen sulfide protects against ventilator-induced lung injury [J]. Anesthesiology, 2010, 113(1): 104-115.
[12] Lee H M, Greeley G H, Herndon D N,etal. A rat model of smoke inhalation injury: influence of combustion smoke on gene expression in the brain [J]. Toxicol Appl Pharmacol, 2005, 208(3): 255-265.
[13] Huang P S, Tang G J, Chen C H,etal. Whole-body moderate hypothermia confers protection from wood smoke-induced acute lung injury in rats: the therapeutic window [J]. Crit Care Med, 2006, 34(4): 1160-1167.
[14] Zou Y Y, Lu J, Poon D J,etal. Combustion smoke exposure induces up-regulated expression of vascular endothelial growth factor, aquaporin 4, nitric oxide synthases and vascular permeability in the retina of adult rats [J]. Neuroscience, 2009, 160(3): 698-709.
[15] Rehberg S, Maybauer M O, Enkhbaatar P,etal. Pathophysiology, management and treatment of smoke inhalation injury [J]. Expert Rev Respir Med, 2009, 3(3): 283-297.
[16] Enkhbaatar P, Traber D L. Pathophysiology of acute lung injury in combined burn and smoke inhalation injury [J]. Clin Sci (Lond), 2004, 107(2): 137-143.
[17] Cox R A, Jacob S, Oliveras G,etal. Pulmonary expression of nitric oxide synthase isoforms in sheep with smoke inhalation and burn injury [J]. Exp Lung Res, 2009, 35(2): 104-118.
[18] Westphal M, Enkhbaatar P, Schmalstieg F C,etal. Neuronal nitric oxide synthase inhibition attenuates cardiopulmonary dysfunctions after combined burn and smoke inhalation injury in sheep [J]. Crit Care Med, 2008, 36(4): 1196-1204.
[19] Rehberg S, Maybauer M O, Maybauer D M,etal. The role of nitric oxide and reactive nitrogen species in experimental ARDS [J]. Front Biosci (Schol Ed), 2010, 2: 18-29.
[20] Westphal M, Rehberg S, Maybauer M O,etal. Cardiopulmonary effects of low-dose arginine vasopressin in ovine acute lung injury [J]. Crit Care Med, 2011, 39(2): 357-363.
[21] Calvert J W. The summer of hydrogen sulfide: highlights from two international conferences [J]. Med Gas Res, 2013, 3(1): 5-9.
[22] Gadalla M M, Snyder S H. Hydrogen sulfide as a gasotransmitter [J]. J Neurochem, 2010, 113(1): 14-26.
[23] Liu W L, Liu Z W, Li T S,etal. Hydrogen sulfide donor regulates alveolar epithelial cell apoptosis in rats with acute lung injury [J]. Chin Med J (Engl), 2013, 126(3): 494-499.
[24] Oh G S, Pae H O, Lee B S,etal. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide [J]. Free Radic Biol Med, 2006, 41(1): 106-119.
[25] Janssen-Heininger Y M, Poynter M E, Baeuerle P A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB [J]. Free Radic Biol Med, 2000, 28(9): 1317-1327.
[26] Zhi L, Ang D, Zhang H,etal. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway [J]. J Leukoc Biol, 2007, 81(5): 1322-1332.
[27] Jeong S O, Pae H O, Oh G S,etal. Hydrogen sulfide potentiates interleukin-1beta-induced nitric oxide production via enhancement of extracellular signal-regulated kinase activation in rat vascular smooth muscle cells [J]. Biochem Biophys Res Commun, 2006, 345(3): 938-944.
[28] Hu L F, Wong P T, Moore P K,etal. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia [J]. J Neurochem, 2007, 100(4): 1121-1128.
[29] Paul B D, Snyder S H. H2S signalling through protein sulfhydration and beyond [J]. Nat Rev Mol Cell Biol, 2012, 13(8): 499-507.
[30] Sen N, Paul B D, Gadalla M M,etal. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions [J]. Mol Cell, 2012, 45(1): 13-24.
[31] LaLonde C, Nayak U, Hennigan J,etal. Plasma catalase and glutathione levels are decreased in response to inhalation injury [J]. J Burn Care Rehabil, 1997, 18(6): 515-519.
[32] Whiteman M, Armstrong J S, Chu S H,etal. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite 'scavenger'? [J]. J Neurochem, 2004, 90(3): 765-768.
[33] Whiteman M, Cheung N S, Zhu Y Z,etal. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? [J]. Biochem Biophys Res Commun, 2005, 326(4): 794-798.
[34] Mustafa A K, Gadalla M M, Suyder S H. Signaling by gasotransmitters[J]. Sci Signal, 2009,2(68):re2.
[35] Liu W, Wang D, Liu K,etal. Nrfz as a converging node for cellular signaling pathways of gasotransmitters[J]. Med Hypotheses, 2012,79(3):308-310.
(2014-01-23收稿 2014-03-02修回)
(责任编辑 岳建华)
Inhaledhydrogensulfideinhibitsoxidativestressofcottonsmokeinhalation-inducedacutelunginjuryinrats
JIANG Yi1,2, HAN Zhihai3, WANG Xiaoyang1, FANG Tingzheng1, HUANG Yan1,and DUAN Yunyou1. 1. Department of Respiratory Medicine, Clinical Medical College of Navy, Second Military Medical University, Beijing 100048, China; 2. Political Department Clinic of Shenyang Military Area Command, Shenyang 110032, China; 3. Pulmonary and Critical Care Medicine of PLA Navy General Hospital, Beijing 100048, China
ObjectiveTo investigate the mechanisms of inhaled hydrogen sulfide inhibiting oxidative stress of cotton smoke inhalation-induced acute lung injury in rats.MethodsTwenty-four male SD rats were randomly allocated into control group, H2S group, smoke group and smoke+H2S group. The rat model of cotton smoke inhalation injury was established. After smoke inhalation or simulated smoke inhalation, rats inhaled H2S 80 ppm, 30% oxygen for 6 hours (H2S group and smoke+H2S group), or rats inhaled 30% oxygen for 6 hours (control group and smoke group). Then rats were mercy killed. In each group of rats we observed the concentration of NO,iNOS,NF-κB p65,MDA in homogenized lung tissue by ELISA,used the method of fluorescence quantitative PCR to detect the expression of iNOS mRNA in homogenized lung tissue, and immunohistochemically detected the relative expression of NF-κB p65 with Image Pro Plus 6.0 software.ResultsCompared with the control group, concentrations of MDA, NO, iNOS, NF-κB p65, relative expression of iNOS mRNA and sum IOD of NF-κB p65 in the smoke group rats’ homogenized lung tissue were significantly elevated, and those in the smoke+H2S group were relatively lower, for example, concentrations of NF-κB p65 were (8123.51±2095.33) pg/ml vs (13803.19±2196.37) pg/ml,P<0.001; relative expression of iNOS mRNA was (1.04±0.24)vs(2.20±0.21),P<0.001. Concentrations of iNOS, relative expression of iNOS mRNA and sum IOD of NF-κB p65 in the H2S group were higher than those in the control group, meanwhile concentrations of MDA, NO, NF-κB p65 in the H2S group were similar to those in the control group.ConclusionsThe mechanisms of inhaled 80 ppm hydrogen sulfide for 6 hours protecting against cottn smoke inhalation-induced ALI in rats potentially is inhaled hydrogen sulfide inhibiting the activation of NF-κB p65, so the expression of iNOS mRNA, iNOS and NO grow downwards and as a result, it relieves oxidative stress and reduces pathological damage to lung tissue.
acute lung injury;smoke inhalation injury;oxidative stress;hydrogen sulfide
全军医学科研“十二五”计划课题(CWS11J180)
姜 毅,博士,主治医师,E-mail:jiangyi1974@sohu.com
1. 100048北京,第二军医大学海军临床医学院呼吸内科;2. 110032沈阳, 沈阳军区政治部门诊部;3. 100048北京,海军总医院呼吸内科
通迅作者:段蕴铀,E-mail:duanyunyou@126.com
R363.274
