Mo-V-Nb多组元氧化物催化乳酸氧化脱氢反应制备丙酮酸
2014-07-14苗诗轩李学兵邢立淑李明时
苗诗轩 宋 亮 李 红 李学兵*, 邢立淑 李明时
(1常州大学石油化工学院,常州 213164)
(2中国科学院生物燃料重点实验室,青岛生物能源与过程研究所,青岛 266101)
0 前 言
丙酮酸是α-酮酸中最简单的分子结构,是最小的具有羰基和羧基两种官能团的分子,显现出特殊的化学性质,常用做药物合成和化工合成的中间体[1]。目前丙酮酸的工业化生产工艺是酒石酸脱水脱羧法[2],而酒石酸是一种石油基原料且近年来价格不断上涨,这限制了丙酮酸的产量,阻碍其商业上的大规模应用。乳酸是公认的三大有机酸之一,目前乳酸的生产工艺是生物发酵法,且随着发酵工艺的不断完善,生产成本逐渐降低[3],因此通过催化乳酸发生氧化脱氢反应将α-羟基氧化成羰基来合成丙酮酸是一种生产丙酮酸的新工艺[4]。
目前,对乳酸及乳酸盐氧化脱氢反应催化体系的研究较不完善,Sugiyama等研究了在水相氧气高压作用下,利用贵金属Pd/C催化剂完成了乳酸盐的氧化脱氢反应合成丙酮酸盐[5],钒催化剂也常被用于选择性氧化脱氢反应[6-7],比如VOCl3催化剂催化乳酸可获得较高的丙酮酸收率,但是VOCl3作为均相催化剂限制了其大规模应用[8],Ai等研究了FePO4催化剂作用下的氧化脱氢反应,利用Fe3+/Fe2+之间的变价,可将乳酸催化转化成丙酮酸,但是同时副反应产物较多[9]。Mo-V-Nb多组元氧化物催化剂常被用于低碳烷烃的选择性氧化脱氢反应,用V、Nb改性后获得的Mo-V-Nb多组元氧化物催化剂具有较好的氧化脱氢反应活性,通常认为,Mo-V-Nb多组元氧化物中的V5+位置上可发生C-H键的活化[10],Mo和Nb共同促进V5+活性中心的再恢复,提高氧化脱氢反应的选择性[11],Mo起稳定骨架结构的作用的同时还可以抑制V5+物种的氧化活性,降低过氧化反应发生的几率,Nb显著影响催化剂的晶化程度,使催化剂颗粒更加细小和无序,并且阻碍会导致碳碳键断裂的单金属氧化物的生成[12-13]。在前期研究的基础上[14],本文用溶剂热合成法合成了对氧化脱氢反应选择性最高的钼钒原子比为2∶1的Mo-V-Nb多组元氧化物 Mo0.61V0.31Nb0.08Ox,采用固定床氧化脱氢反应评价装置,探索了乳酸氧化脱氢反应过程,分析了反应温度、氧分压、质量空速对丙酮酸选择性和乳酸转化率的影响,并研究了基于该催化剂乳酸氧化脱氢可能的反应路径。
1 实验部分
1.1 催化剂的制备
采用前期研究所用的溶剂热合成法来合成钼钒原子比为2∶1的Mo-V-Nb多组元氧化物Mo0.61V0.31Nb0.08Ox。先 将 10.77 g (NH4)6Mo7O24·4H2O(Aladdin,99.9% ,metals basis)、2.42 g C4H4NNbO9·xH2O (Sigma,99.99% ,trace metals basis) 和 7.2 g C2H2O4·2H2O(Aladdin,优级纯)分别溶于 25 mL 纯水中制成溶液,再将 3.63 g NH4VO3(Aladdin,99.9%,metals basis)溶于 C2H2O4·2H2O溶液中,然后在搅拌过程中依次将 (NH4)6Mo7O24·4H2O溶液和C4H4NNbO9·xH2O 溶液加入 NH4VO3与 C2H2O4·2H2O的混合溶液中,升温至90℃搅拌蒸干溶液,随后移入烘箱中120℃烘干过夜,最后将烘干后的粉末在空气气流中400℃焙烧4 h制得催化剂,焙烧时空气流速 100 mL·min-1,升温速率为 10 ℃·min-1。
1.2 催化剂的表征
采用德国布鲁克公司的Bruker AXS-D8型X射线衍射光谱仪 (X-ray diffraction,XRD)来分析Mo0.61V0.31Nb0.08Ox催化剂的晶相,Cu-Kα 靶射线,管电压 40 KV,电流 40 mA,扫描范围 2θ=10°~60°,步长0.02°。
采用日本Hitachi公司的S-4800型扫描电子显微镜 (Scanning Electron Microscope,SEM)来表征Mo0.61V0.31Nb0.08Ox催化剂的表面形貌,加速电压 10.0 kV,放大倍数10万倍,并用EDS检测催化剂表面各元素的分布和比例。
2.3 催化剂的活性评测
Mo0.61V0.31Nb0.08Ox催化剂上乳酸氧化脱氢反应活性评价在常压连续流动固定床反应器中进行,氦气(99.999%,Air Liquid)和氧气(99.999%,合利公司)分别流经过滤器、截止阀、质量流量计和单向阀后与DL-乳酸(Sigma,90%)混合进入石英反应管(内径 1.0 cm,长度40 cm)中,气体流量由EL-Flow型质量流量计(荷兰Bronkhorst High-Tech公司)控制,乳酸进样速度由NE-1600型微量注射泵(美国Era公司)控制。Mo0.61V0.31Nb0.08Ox催化剂(60-80 目)与等质量的石英砂(60-80目)混合后由适量石英棉固定在石英反应管中,每次反应装填催化剂时都确保催化剂处于管式炉的恒温段内,在反应进行1 h(此时反应已达到稳态)后开始取样,产物收集装置用冰水浴冷却。使用 GC-MS(Agilent 7890A-Agilent 5975C)对产物进行定性分析,INNOWax 色谱柱 (HP,30 m×0.32 mm)。使用气相色谱(Varian 450GC)对产物进行定量分析,FID 检测器,INNOWax 色谱柱(HP,30 m×0.32 mm)。乳酸转化率和各产物的选择性是通过将各产物的碳数归一化后进行相应计算得出的[15]。
2 结果分析与讨论
2.1 催化剂的表征结果
Mo0.61V0.31Nb0.08Ox催化剂的 XRD 图见图 1。如图所示,Mo0.61V0.31Nb0.08Ox催化剂在 2θ为 21.94°、24.78°和33.64°附近有3个强峰,经分析这些衍射峰是[Mo(V,Nb)]5O14(JCPDS No.58-0788)的 衍 射 峰[16-17],除了这 3 个强峰以外,Mo0.61V0.31Nb0.08Ox催化剂在 2θ为23.34°、27.34°和 33.27°附近还有 3 个强峰,经分析这些衍射峰是α-MoO3(JCPDS No.76-1003)的衍射峰,除此之外,从图中还可以看出,Mo0.61V0.31Nb0.08Ox催化剂在2θ为20°到30°之间存在1个宽峰,从这可以推断出Mo0.61V0.31Nb0.08Ox催化剂中还有部分无定形态物质的存在。
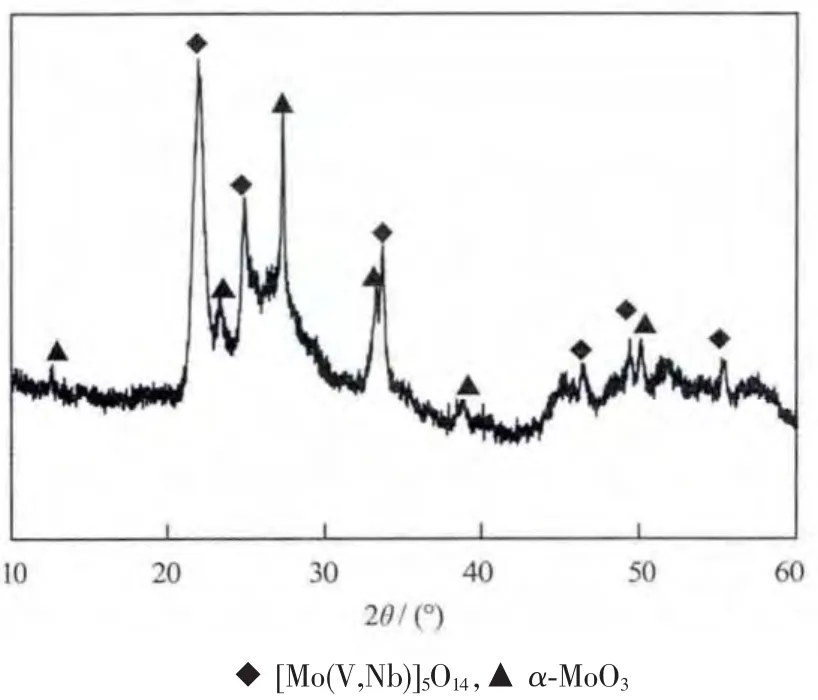
图1 Mo0.61V0.31Nb0.08Ox催化剂的XRD图Fig.1 Powder X-ray diffraction pattern of Mo0.61V0.31Nb0.08Ox
图2为Mo0.61V0.31Nb0.08Ox催化剂相同测试条件下不同区域的SEM图像,如图所示,Mo0.61V0.31Nb0.08Ox催化剂中存在2种明显不同的组分,一种是不规则的颗粒状物质,如图2(a),经过对比分析认为这些不规则的颗粒状物质是活性组分[Mo(V,Nb)]5O14[17-18],另一种物质呈现出片状结构,如图2(b),经EDS分析主要元素为Mo和O,结合XRD的物相分析结果认为片状结构为α-MoO3。
目前,普遍认为[Mo(V,Nb)]5O14是Mo-V-Nb多组元氧化物催化剂的活性相,并且是结晶度较低的更接近于无定形态的[Mo(V,Nb)]5O14才具有良好的催化活性[10,18]。根据现有的文献报道[10-14],目前Mo-V-Nb多组元氧化物催化剂的合成方法主要有溶剂热合成法和水热法,水热法可以合成单一的[Mo(V,Nb)]5O14结构,但是这种[Mo(V,Nb)]5O14结晶度较好,有效的反应比表面较低,不具有良好的催化活性,因此主要用于进行Mo-V-Nb多组元氧化物催化剂的结构分析[19],溶剂热合成法可以合成结晶度低的更接近无定形态的[Mo(V,Nb)]5O14结构,但是催化剂中一般含有部分由未能进入[Mo(V,Nb)]5O14结构的钼或者钒形成的氧化物[13,18]。结合催化剂的表征结果来看,本文通过溶剂热合成法合成的Mo0.61V0.31Nb0.08Ox催化剂中含有活性组分[Mo(V,Nb)]5O14,除此之外,还有部分由未能进入[Mo(V,Nb)]5O14结构的钼形成的α-MoO3。

图2 (a)[Mo(V,Nb)]5O14组分的SEM图像和 (b)α-MoO3组分的SEM图像Fig.2 SEM images of(a)[Mo(V,Nb)]5O14and(b)α-MoO3
2.2 反应产物的分析结果
用GC-MS分析乳酸氧化脱氢反应的产物,发现产物中存在未反应完的DL-乳酸、丙酮酸、乙酸、丙烯酸、柠康酸酐(GC-MS给出的结果是柠康酸酐或者衣康酸酐,以下统一以柠康酸酐计算)和2-甲基琥珀酸酐。推测的反应路径如图3所示,在Mo0.61V0.31Nb0.08Ox的催化作用下,部分乳酸发生氧化脱氢反应生成丙酮酸,部分乳酸发生分子内脱水反应生成丙烯酸,部分生成的丙烯酸和丙酮酸会发生过氧化反应以致碳链断裂生成乙酸和二氧化碳,还有少部分乳酸与生成的乙酸发生反应生成2-甲基琥珀酸酐,部分2-甲基琥珀酸酐会发生氧化脱氢反应生成柠康酸酐,但是生成的丙烯酸、柠康酸酐和2-甲基琥珀酸酐的量极少(小于1%)。
2.3 反应温度

图3 乳酸氧化脱氢推测的反应路径图Fig.3 Plausible reaction pathway of lactic acid oxidative dehydrogenation over Mo-V-Nb multi-component oxides

图4 温度对反应的影响Fig.4 Effect of temperature on the oxidative dehydrogenation of lactic acid
图4为反应温度对Mo0.61V0.31Nb0.08Ox催化剂催化性能的影响,如图所示,在所选定的温度范围内,随着反应温度的升高,乳酸的转化率和乙酸的选择性不断上升,丙酮酸的选择性不断下降,收率先上升后下降,这和预想的结果一致。乳酸在反应温度超过250℃以后趋近于完全转化,这是因为随着反应温度的升高,反应速率也不断提高,在不改变接触时间的情况下会有更多的乳酸被活化发生反应。反应温度低于190℃时丙酮酸的选择性在90%以上,但升高反应温度后,丙酮酸的选择性急剧降低,乙酸和二氧化碳的选择性却急剧升高,这是因为乳酸、丙烯酸和丙酮酸碳链断裂的活化能要高于乳酸氧化脱氢和脱水的活化能,因此在反应温度升高的情况下,反应更倾向于向活化能高的方向进行。由此可见,乳酸氧化脱氢合成丙酮酸的生产工艺可行,但是需要控制反应温度,因为过高的反应温度虽然会使转化率升高,但是不利于反应向生成丙酮酸的方向进行,生成副产物乙酸和二氧化碳,原子经济性不高,不利于实际生产过程中的循环生产。
2.4 氧气分压
通过调整氧气和氦气的流速来控制反应时氧气的分压。图5为氧气分压对Mo0.61V0.31Nb0.08Ox催化剂催化性能的影响,如图所示,在所选择的氧气分压范围内,随着氧气分压的升高,乳酸的转化率也在升高,并且在氧分压大于38 kPa以后,上升趋势趋于平缓,这是由于随着氧分压的增加,当氧气相对于反应物过量后,其对于转化率的影响逐渐减小。氧气分压对丙酮酸选择性的影响很小,在所选择的氧气分压范围内,仅下降了2%,乙酸和二氧化碳的选择性也仅仅上升了不到1%,这可能是由于氧气分压升高导致催化剂表面产物脱附较以前困难以致部分产物过氧化造成的,由此可以看出,氧气被活化是反应的快速步骤,反应物在催化剂上的吸附和活化是反应的决速步骤。这四组实验中,氧气分压为20 kPa时,氧气与氦气的分压比为1:4,这是模拟空气中氧气的分压进行实验的,从实验结果可以看出,虽然转化率只有30%,但是选择性高达90%,因此在实际生产过程中,可以将氧化性气体从氧气换为空气,进行串连式循环反应,这样可以节约生产成本。

图5 氧分压对反应的影响Fig.5 Effect of partial pressure of oxygen on the oxidative dehydrogenation of lactic acid
2.5 反应空速
不同的空速通过在不改变其他反应条件的情况下调整催化剂用量进而改变反应物与催化剂的接触时间来实现。图6为根据反应结果做出的转化率与各产物选择性之间的关系图,如图所示,在保持其他反应条件不变的情况下,随着反应时催化剂使用量的增加,乳酸的转化率不断升高,丙酮酸的选择性明显下降,乙酸和二氧化碳的选择性急剧升高,收率不断升高但上升趋势逐渐变平缓最后趋近于一个恒定值,约为50%。将乳酸转化率外推到0,可得乙酸和二氧化碳的初始选择性为0,丙酮酸初始选择性约为100%,还有痕量的丙烯酸。该结果表明,在所选定的实验条件范围内,Mo0.61V0.31Nb0.08Ox催化剂上乳酸的氧化脱氢反应主要存在两个平行竞争反应,氧化脱氢生成丙酮酸和分子内脱水生成丙烯酸,并且绝大多数乳酸倾向于前者,部分生成的2种产物在未能及时从催化剂上脱附的情况下,会发生过氧化反应导致碳链断裂生成乙酸和二氧化碳,或者和催化剂上吸附的乳酸反应生成2-甲基琥珀酸酐,部分未能及时脱附的2-甲基琥珀酸酐又发生氧化脱氢反应生成柠康酸酐。
3 结 论
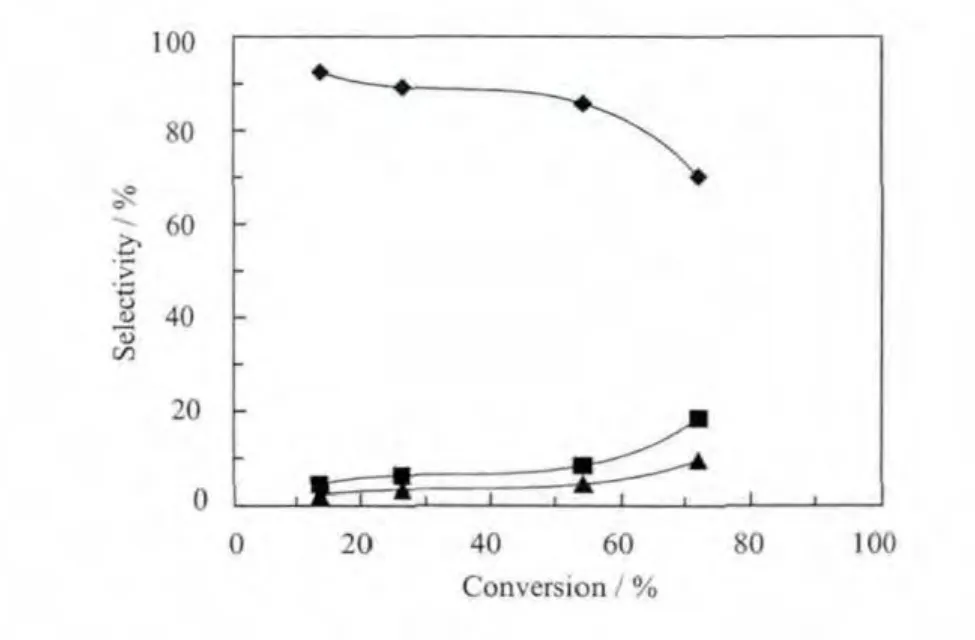
图6 空速对反应的影响Fig.6 Effect of space velocity on the oxidative dehydrogenation of lactic acid
本文采用Mo-V-Nb多组元氧化物为催化剂催化乳酸氧化脱氢合成丙酮酸,经分析,发现Mo-V-Nb多组元氧化物中除了不规则颗粒状的活性组分[Mo(V,Nb)]5O14外还存在部分片状的α-MoO3。产物主要有丙酮酸、乙酸以及少量的丙烯酸、柠康酸酐和2-甲基琥珀酸。反应温度低于190℃时丙酮酸的选择性在90%以上,250℃以上乳酸几乎完全转化,但是丙酮酸的选择性降至20%以下。升高氧分压会促使转化率略微升高,但是对丙酮酸、乙酸和二氧化碳的选择性影响不大。延长反应物与催化剂的接触时间虽然会提升乳酸的转化率,但是丙酮酸的选择性下降,收率逐渐趋于恒定,约为50%。钒铌多组元氧化物催化乳酸氧化脱氢合成丙酮酸,该反应具有能耗低、无污染、经济性高等特点,具有较好的发展前景和商业价值。
[1]Liu Z Q,Jia L Z,Zheng Y G.Process Biochem.,2010,45:1632-1637
[2]Gao C,Ma C Q,Xu P.Biotechnol.Adv.,2011,29:930-939
[3]Fan Y X,Zhou C H,Zhu X H.Cat.Rev.-Sci.Eng.,2009,51(3):293-324
[4]Lomate S,Bonnotte T,Katryniok B,et al.J.Mol.Catal.A:Chem.,2013,377:123-128
[5]Tsyjino T,Ohigashi S,Hayashi H,et al.J.Mol.Catal,1992,71:25-35
[6]LI Kang(李 康),HAN Yu-Wang(韩 毓 旺),GE Xin(葛 欣),et al.Chinese J.Inorg.Chem.(无机化学学报),1999,5(6):826-830
[7]JIANG Qiao(江桥),ZHAO Jing(赵静),JI Wei-Jie(季伟捷),et al.Chinese J.Inorg.Chem.(无机化学学报),2008,24(9):1474-1479
[8]Yasukawa T,Ninomiya W,Mae K,et al.Ind.Eng.Chem.Res.,2011,50:3858-3863
[9]Ai M,Ohdan K.Appl.Catal.A:Gen.,1997,150:13-20
[10]Concepción P,Botella P,López-Nieto J M.Appl.Catal.A:Gen.,2004,278:45-56
[11]Li X B,Iglesia E.Chem.Eur.J.,2007,13(33):9324-9330
[12]Roussel M,Bouchard M,Bordes-Richard E,et al.Catal.Today,2005,99:77-87
[13]López-Medina R,Fierro J L G,Guerrero-Pórez M O,et al.Appl.Catal.A:Gen.,2010,375:55-62
[14]Li X B,Iglesia E.Appl.Catal.A:Gen.,2008,334:339-347
[15]Holm T.J.Chromatogr.A,1999,842:221-227
[16]Kardash T Y,Plyasova L M,Bondareva V M,et al.Appl.Catal.A:Gen.,2010,375:26-36
[17]Sanchez M,Sanchez F,Trunschke A,et al.Angew.Chem.Int.Ed.,2012,51:7194-7197
[18]Roussel M,Bouchard M,Bordes-Richard E,et al.Catal.Today,2005,99:77-87
[19]Zhang W,Trunschke A,Su D S,et al.Angew.Chem.Int.Ed.,2010,49:6084-6089
