8-喹啉氧基乙酸铅(Ⅱ)一维配位聚合物的合成、晶体结构和性质
2014-07-14王玉红宋瑞峰徐孝文
王玉红 宋瑞峰 徐孝文
(1苏州科技学院化学生物与材料工程学院,苏州 215009)
(2江苏省环境功能材料重点实验室,苏州 215009)
近20年来,配位聚合物在多相催化、气体吸附、分离、离子交换等领域显示了潜在的应用,引起了人们极大的兴趣[1-7],但相关工作主要集中在d区过渡金属配位聚合物合成与应用方面[2-3,8-9],主族金属配位聚合物的设计合成相对较少。铅是一种主族有毒的重金属,生物体系中的铅(Ⅱ)模型配合物和配位螯合剂除去铅(Ⅱ)的研究特别受到重视;同时,由于铅(Ⅱ)具有较大的半径,易变的立体化学活性和灵活的配位环境,为新型金属有机框架构建提供了独特的机会,一些铅基配位聚合物相继报道[10-13]。8-喹啉氧基乙酸(H2QOA)是一种多齿N/O供给配体,具有与多金属结合和形成氢键的能力,与d区和镧系金属已构筑出有趣结构的配位聚合物[14-17]。到目前为止,8-喹啉氧基乙酸与铅(Ⅱ)构筑配位聚合物的研究尚未见文献报道。本文以8-喹啉氧基乙酸为配体,首次合成了一个新的一维铅(Ⅱ)配位聚合物1,通过X-射线单晶衍射确定了它的结构,并测定了它的固体紫外漫反射和荧光光谱,表征了其光学性质。
1 实验
1.1 实验试剂及仪器
所用试剂均为分析纯。8-喹啉氧基乙酸按文献[18]的方法制备。
化合物的元素含量在Perkin-Elmer 240C元素分析仪 (美国PERKIN-ELMER公司)测定;红外光谱利用KBr压片法在170 SX (Nicolet)FT-IR光谱仪(美国 NICOLET 公司) 测定 (4 000~400 cm-1);TGADTA热分析在SDT 2960型热分析仪上进行;粉末X射线衍射(PXRD)测定在D/MAX-3C衍射仪上进行,以 Cu Kα(λ=0.154 06 nm)为辐射光源,测试条件为40 kV和30 mA;固态UV-Vis光谱用压片法在Shimaduz公司UV-240紫外可见分光光度计测定;固体荧光光谱在Cary Eclipse荧光光谱仪上测定;晶体衍射数据在Rigaku Mercury CCD X射线单晶衍射仪收集。
1.2 配合物1的合成
在硬质Pyrex玻璃管中加入8-喹啉氧基乙酸(20.3 mg,0.1 mmol)和三乙胺水溶液(0.1 mol·L-1,1.2 mL),振荡溶解,然后加入 Pb(Ac)2·3H2O(41.1 mg,0.1 mmol)和蒸馏水(2.8 mL),封管后放入电热恒温干燥箱中;缓慢升温至150℃,恒温75 h,以每100 min 5℃的速率降至室温。得到适于X-射线结构分析的黄色晶体配合物1,产率:51%。红外光谱数据(KBr压片,cm-1):1 583.8,1 504.2,1 421.6,1 315.2,1 116.6,757.9。元素分析理论值为(C22H16N2O6Pb)(%):C,43.21;H,2.64;N,4.58; 实验值为 (%):C,42.99;H,2.67;N,4.59。
1.3 配合物的晶体结构测定
取大小为 0.40 mm×0.16 mm×0.16 mm 的黄色单晶置于Rigaku Mercury CCD X射线单晶衍射仪上,用石墨单色化的 Mo Kα 射线(λ=0.071 073 nm),在温度为223(2)K下,以φ-ω扫描方式收集衍射数据。在 3.14°<θ<27.46°范围内共收集数据点 5 011个,其中独立衍射点 2 221 个(Rint=0.033 0),I>2σ(I)的可观测的衍射点1 979个。用CrystalClear程序包进行数据还原(Rigaku&MSC,1999)。衍射数据经Lp因子校正和吸收因子校正 (multi-scan),其中对I>2σ(I)衍射点参加结构修正。晶体结构采用SHELXL97程序由直接法解出[19],结构精修采用SHELXL97程序[20],确定氢原子的方法是理论加氢,对氢原子和非氢原子分别采用各向同性和各向异性热参数对结构进行全矩阵最小二乘法修正。最终偏差因子 R1=0.022 9 和 wR2=0.054 1(w=1/[σ2(Fo2)+(0.024 1P)2+0.000P],其中 P=(Fo2+2Fc2)/3)。标题配合物的晶体学数据列于表1,主要键长和键角列于表2。
CCDC:939269。
2 结果与讨论
2.1 配合物1的红外光谱
化合物1的红外光谱如图1所示。1 583.8与1 421.6 cm-1的强吸收峰分别被指派为COO-的不对称吸收峰νas(COO-)和对称吸收峰(νs(COO-))。配合物1中,羧基的特征峰(vC=O1 760 cm-1)和vOH的特征峰(3 200~2 500 cm-1)消失,出现了羧酸盐特有的反对称伸缩振动vas(COO-)和对称伸缩振动vs(COO-)的特征峰,表明配体的羧酸脱掉质子以酸根的形式参与配位。在1 700 cm-1附近没有配体羰基峰出现,表明配体羧基上的质子已全部脱掉,说明了8-喹啉氧基乙酸根离子配位到铅离子后发生了振动位移,根据COO-不对称吸收峰vas(COO-)与对称吸收峰vs(COO-)的数值大小(Δv=vas(COO-)-vs(COO-)=162.2 cm-1),可推测出配合物1中8-喹啉氧基乙酸根离子存在双齿螯合配位模式[21]。

表1 配合物1的晶体数据和结构修正参数Table 1 Crystal data and structure refinements for the complex 1

表2 配合物1的主要键长和键角Table 2 Selected bond lengths(nm)and angles(°)for the complex 1

图1 配合物1的红外光谱Fig.1 Infrared spectrum of the complex 1
2.2 配合物1的晶体结构
配合物1为一维链状配位聚合物。图2为配位聚合物1的分子结构图,一维链状结构见图3。配合物1中,Pb2+采取六配位的几何构型,每个Pb(Ⅱ)离子与4个QOA配体上的6个羧基氧原子配位,其中2个QOA配体上的羧基以双齿螯合配位 (O1和O2、O1i和O2i),剩余2个QOA配体各以1个羧基氧原子(O1ii和O1iii)配位。相邻的2个Pb(Ⅱ)离子通过2个QOA配体上羧基氧的μ2-O原子桥联作用,沿a轴方向连接成一维链状结构配位聚合物 (图3),相邻的Pb与Pb之间的距离为0.450 03 nm。围绕Pb(1)离子的键角从 49.62(8)°变化到 176.25(12)°。键角 O1iii-Pb1-O1ii大小为 176.25(12)°,表明 O1iii、Pb1 与 O1ii近似在同一直线上。在Pb(Ⅱ)离子配位环境中,所有配位氧原子均在以Pb1离子为中心球体的一侧,因此Pb2+是典型的半定向六配位模式。如果考虑Pb(Ⅱ)离子与喹啉氮原子的弱配位作用 (Pb…N距离为0.289 8 nm),Pb2+则为典型的全定向七配位模式。

图2 配合物1的分子结构图Fig.2 A view of molecular structure of the complex 1

图3 配合物1的一维链状结构Fig.3 Perspective view of the 1D chain structure of the complex 1
在配位聚合物1中,每个去质子化的QOA配体的羧基,不仅作为Pb(Ⅱ)离子的双齿配体,也是相邻Pb(Ⅱ)离子的单齿配体。其中,桥联原子O1、O1ii和O1i、O1iii分别把相邻的 Pb1、Pb1ii和 Pb1、Pb1i连接起来,形成Pb1-O1-Pb1ii-O1ii和Pb1-O1i-Pb1i-O1iii2个四元环Pb2O2,构成一维链状结构,如图3所示。Pb1-O1i与 Pb1i-O1iii的键长为 0.259 9(3)nm,Pb1-O1iii与 Pb1i-O1i的键 长为 0.268 2(3)nm;O1i-Pb1-O1iii和 O1i-Pb1i-O1iii的键角 63.12(10)°,Pb1-O1i-Pb1i和 Pb1i-O1iii-Pb1 的键角为 116.88(9)°,说明 Pb1、O1iii、Pb1i和 O1i组成了 1个平行四边形 Pb2O2。所有的对等平行四边形Pb2O2在Pb(Ⅱ)离子的连接下无限延伸。桥连的羧基氧原子的键长为O1-Pb1 0.259 9(3)nm、O1-Pb1ii0.268 2(3)nm,平均键长小于非桥连的羧基氧原子键长O2-Pb1 0.268 2(3)nm,结果与文献报道的双氧原子桥连形成的Pb2O2环一致[22-25]。
2.3 配合物1的UV-Vis-NIR漫反射光谱
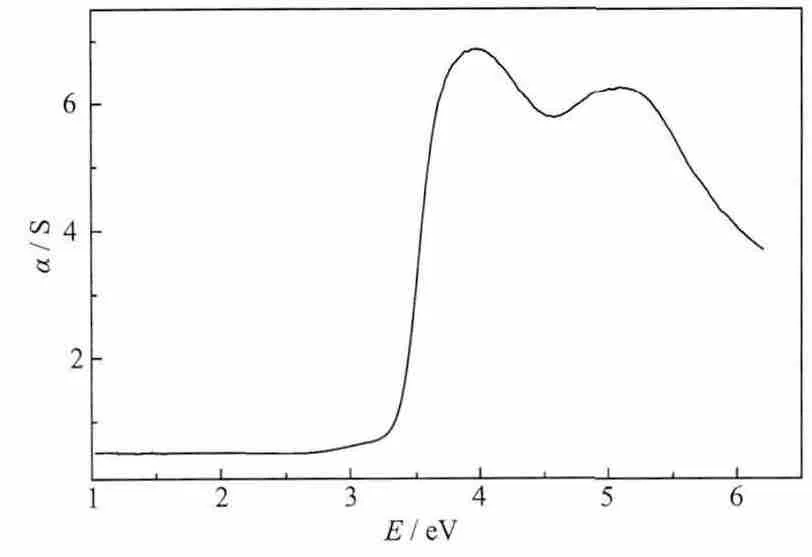
图4 配合物1的紫外可见漫反射光谱Fig.4 Optical diffuse reflecctance spectrum of the complex 1
测定了配位聚合物1的UV-Vis-NIR漫反射光谱,根据UV-Vis-NIR漫反射光谱数据,计算入射光子能量E对应的Kubelka-Munk函数,即F=α/S=(1-R∞)2/R∞[26],式中,R∞是对试样为无限厚(透射为零)时的反射率。将K-M函数F对入射光的能量E(eV)作图,得到了配位聚合物1的F-M函数对E(eV)的曲线(图4)。配合物1的光学能隙约为3.4 eV,表明配合物1具有潜在的半导体性质。
2.4 配合物1的固体荧光光谱
图5为室温在350 nm波长的光激发下,测定的配合物1和配体的固体荧光光谱。从图中可以看出,配体的固体荧光光谱归因于喹啉基团的荧光发射峰。配合物1在397 nm附近出现一个窄的荧光发射最高峰,于600~670 nm范围出现了一个宽的发射峰。前者可归因于配体到金属的荷移跃迁,进一步表明了喹啉基团与Pb(Ⅱ)的弱配位作用;后者可归因于[Pb2(μ-O)2]作用,与UV-Vis-NIR漫反射光谱一致。

图5 配合物1与配体的荧光光谱Fig.5 Fluorescence spectra of the complex 1 and the ligand QOAH
2.5 配合物1的粉末X-射线和热稳定性分析

图6 (a)配合物1的粉末X-射线图(实验值与模拟值),(b)配合物1的TGA-DTA热分析曲线Fig.6 (a)Experimental and simulated power X-ray diraction patterns of the complex 1,(b)TGA-DTA curves of the complex 1
如图6a所示,粉末X-射线的实验结果表明配合物1的实验值和晶体数据拟合值较好地吻合,说明合成得到的大量样品是纯样品。通过TGA-DTA热分析研究了化合物1的热稳定性(图6b),在30~800℃温度范围内,氮气流为100 mL·min-1,以升温速率为10℃·min-1研究了配合物1的失重情况。在294℃以前,配合物1是稳定的。继续升温,开始失重,失重速率快,294~800℃温度范围内失重了 54.2%,在302.5℃时观察到DTA曲线上存在1个吸热峰。45.8%的残余物应为PbO和单质碳的混合物。
[1]Kitagawa S,Kitaura R,Noro S I.Angew.Chem.Int.Ed.,2004,43:2334-2375
[2]DU Mao(杜淼),BU Xian-He(卜显和).Progress in Chemistry(化学进展),2009,21:2458-2464
[3]Férey G,Serre C.Chem.Soc.Rev.,2009,38:1380-1399
[4]Takashi U,Nobuhiro Y,Susumu K.Chem.Soc.Rev.,2009,38:1228-1236
[5]Deng H,Doonan C J,Furukawa H,et al.Science,2010,327:846-850
[6]Ohara K,Kawano M,Inokuma Y,et al.J.Am.Chem.Soc.,2010,132:30-31
[7]Xie Z G,Ma L Q,Kathryn E,et al.J.Am.Chem.Soc.,2010,132:922-923
[8]Min K S,Suh M P.Chem.Eur.J.,2001,7:303-313
[9]Bradshaw D,Prior T J,Cussen E J,et al.J.Am.Chem.Soc.,2004,126:6106-6114
[10]Yang J,Li G D,Cao J J,et al.Chem.Eur.J.,2007,13:3248-3261
[11]Fan S R,Zhu L G.Inorg.Chem.,2007,46:6785-6793
[12]Yang J,Ma J F,Liu Y Y,et al.Inorg.Chem.,2007,46:6542-6555
[13]Zhang L,Qin Y Y,Li Z J,et al.Inorg.Chem.,2008,47:8286-8293
[14]Wang Y H,Song R F,Zhang F Y.J.Mol.Struct.,2005,752:104-109
[15]Cheng X N,Zhang W X,Chen X M.J.Am.Chem.Soc.,2007,129:15738-15739
[16]Fan J,Wang Z H,Yin X,et al.CrystEngComm,2010,12:216-225
[17]WANG Yu-Hong(王玉红),ZHAO Kai-Yan(赵凯元),DU Juan(杜鹃).Chinese J.Inorg.Chem.(无机化学学报),2005,21:511-514
[18]Koelsch C F.J.Am.Chem.Soc.,1931,53:304-305
[19]Sheldrick G M.SHELXS-97,Program for X-ray Crystal Structure Solution,Germany,University of Göttingen,1997.
[20]Sheldrick G M.SHELXL-97,Program for X-ray Crystal Structure Refinement,Germany,University of Göttingen,1997.
[21]Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordination Compounds,6th Ed.NewYork:John Wiley&Sons,2009.
[22]Foreman M R St J,Plater M J,Skakle J M S.J.Chem.Soc.,Dalton Trans.,2001:1897-1903
[23]Zhao Y H,Xu H B,Fu Y M,et al.Cryst.Growth Des.,2008,8:3566-3576
[24]Foreman M R St J,Gelbrick T,Hursthouse M B,et al.Inorg.Chem.Commun.,2000,3:234-238
[25]Shahverdizadeh G H,Soudi A A,Morsali A,et al.Inorg.Chim.Acta,2008,361:1875-1884
[26]Wendlandt W W,Heeht H G.Reflectance Spectroscopy.New York:Interscienee Publishers,1966.
