离子热合成形貌可控的羟基磷灰石超细粉体
2014-07-14刘慧慧孔爱国王海文
殷 馨 刘慧慧 孔爱国 王海文*,
(1华东理工大学化学与分子工程学院,上海 200237)
(2华东师范大学化学系,上海 200241)
0 引 言
羟基磷灰石(Hydroxyapatite,简称HAP或HA),分子式为Ca10(PO4)6(OH)2,是人体和动物骨骼的主要无机成分[1],其在骨质中含量约为60%,是一种长度为200~400 nm,厚度15~30 nm的针状结晶,周围规则地排列着骨胶原纤维。医学领域长期以来广泛使用的是金属有机高分子等生物医学材料,其生物兼容性和人体适应性并不令人满意。因此,近二十年来,研究接近或类似于自然骨成分的无机生物医学材料极其活跃,其中特别值得重视的是对羟基磷灰石材料的研究。因为羟基磷灰石是一种典型的生物材料,具有优良的生物相容性和生物活性,是理想的骨组织替代材料[2]。所以近来纳米羟基磷灰石的制备方法及医学应用的研究已越来越受到研究工作者的广泛关注。
室温离子液体给化学研究开辟了一个全新的领域,它不仅在电化学[3]、有机合成[4-6]、催化[7-9]、分离[10-13]等方面具有独特的作用,而且伴随着绿色化学概念的提出,室温离子液体有望为面临全球环境安全问题的现代工业带来突破性的发展。2004年,Cooper等首次在咪唑类离子液体中合成了一系列微孔磷酸铝分子筛,并提出了离子热合成法这个概念[14]。由于离子液体几乎没有蒸汽压,与传统的水热和溶剂热合成方法相比,这种新的方法可以减少大量挥发性有机溶剂,将纳米材料的合成带到了一个完全崭新的领域,因而受到人们的高度重视。最近几年离子热合成法在纳米材料制备方面展现出广阔的应用前景。Hideaki等[15]在咪唑类离子液体中合成出了纳米级的金微粒。Ki-Sub等[16]用硫羟基取代的新型离子液体为稳定剂,制备了纳米级的金和铂微粒。
本文中我们利用离子液体蒸汽压低且可重复利用的优点,提出了一种新的无需高温焙烧制备羟基磷灰石超细粉体的方法,其中,离子液体既起到溶剂的作用又起到修饰剂的作用,通过调整离子液体的阳离子和阴离子,可以调整所合成的羟基磷灰石的形貌。本文中使用的离子液体为烷基咪唑类的离子液体,具体为 BmimBr,BmimBF4,EmimBr,EmimBF4。与普通沉淀法所得到的羟基磷灰石相比,本文制备合成的羟基磷灰石对有机染料碱性品红具有较好的吸附性能。
1 实验部分
在制备合成羟基磷灰石超细粉体时所用到的化学药品均为分析纯,并由中国医药集团上海化学试剂公司提供。
1.1 室温离子液体的制备
本实验所用4种室温离子液体溴化1-甲基-3-丁基咪唑 (BmimBr),1-甲基-3-丁基咪唑四氟硼酸盐(BmimBF4)及 1-甲基-3-乙基咪唑(EmimBr),1-甲基-3-乙基咪唑四氟硼酸盐(EmimBF4)的制备方法如下[17-18]:
(1)BmimBr
在一配有滴液漏斗、回流冷凝器、氮气通入管、磁力搅拌器的250 mL四口圆底烧瓶中,加入20 mL(约0.25 mol)1-甲基咪唑和80 mL甲苯溶液,在N2保护下,通过滴液漏斗向其中滴入(约20 min)30 mL(约0.28 mol)的溴代正丁烷,混合物在搅拌下加热回流(约70℃),维持此温度回流48 h。然后将反应混合物冷却。分出上层的甲苯层,剩余液体用50 mL的乙酸乙酯洗涤2次,在洗涤过程中就有大量的白色晶体析出,倾出反应容器中的液体,将固体转移到真空干燥箱中抽真空,将其中的溶剂全部抽去后白色固体用20 mL乙腈和40 mL乙酸乙酯重结晶,得纯白色固体,即为BmimBr。
(2)BmimBF4
在一配有干燥管、N2通入管、磁力搅拌器的250 mL圆底烧瓶中加入50 mL丙酮,在N2保护下,加入 19.1 g(0.1 mol)溴化 1-甲基-3-丁基咪唑和10.48 g(0.1 mol)NaBF4固体,常温电磁搅拌24 h,然后将反应混合物过滤,滤液旋转蒸发除去丙酮,然后放于真空干燥箱中干燥24 h。得约12 mL无色液体即BmimBF4。
(3)EmimBr
在一配有滴液漏斗、回流冷凝器、N2通入管、磁力搅拌器的250 mL四口圆底烧瓶中,加入20 mL(约0.25 mol)1-甲基咪唑和80 mL甲苯溶液,在N2保护下,通过滴液漏斗向其中滴入(约30 min)56 mL(约0.75 mol)的溴乙烷,在剧烈搅拌下,回流48 h。然后冷却至室温,可以看到液体分层,分出上层的甲苯,用50 mL的甲苯和50 mL的乙酸乙酯依次对下层淡黄色液体进行洗涤,洗涤3次,将得到的淡黄色液体密封,放入冰箱中冷却过夜,即可得到黄白色结晶。将此晶体溶于20 mL沸腾的乙腈中,加入乙酸乙酯30 mL进行重结晶,此重结晶过程进行2次以上,直到晶体为纯白色为止。将此晶体置于真空干燥箱中抽真空3 h,除去其中的挥发性成分,得到白色固体,即为EmimBr。
(4)EmimBF4
在一配有干燥管、N2通入管、磁力搅拌器的250 mL圆底烧瓶中加入50 mL丙酮,在N2保护下,将19.1 g(0.1 mol)溴化1-甲基-3-乙基咪唑和 10.48 g(0.1 mol)NaBF4固体加入,常温电磁搅拌24 h,然后将反应混合物过滤,滤液旋转蒸发除去丙酮,然后放于真空干燥箱中干燥24 h。得12 mL无色液体即EmimBF4。
1.2 羟基磷灰石超细粉体的制备
首先将0.200 g(2.699 mmol)氢氧化钙和0.186 g(1.613 mmol)磷酸,溶于2 g离子液体并置于反应釜中,室温搅拌使之充分混匀后,将反应釜置于120℃的烘箱中反应72 h。反应完毕室温冷却后过滤所得到的白色固体沉淀,经洗涤,烘干,即可得到羟基磷灰石超细粉体。合成方法中离子液体指BmimBr,BmimBF4,EmimBr,EmimBF4。
1.3 羟基磷灰石对低浓度碱性品红水溶液的吸附实验
实验在中性条件(pH=7)下进行。将有机染料碱性品红溶于水,配成200 mg·L-1的溶液。分别取10 mg BmimBr为溶剂所合成的羟基磷灰石和水为溶剂所合成的羟基磷灰石置于10 ml 200 mg·L-1的碱性品红溶液中,以相同的搅拌速率进行搅拌。搅拌一定时间后,将固体粉末离心分离,取上层清液以721型分光光度计在544 nm处测定碱性品红的吸光度值,计算吸附量。
1.4 测试表征
采用Rigaku公司D/MAX 2200PC X-射线粉末衍射进行样品的物相分析,使用Cu Kα为射线源仪(λ=0.154 06 nm),石墨单色器。高角度物相分析的管电压和电流为40 kV和40 mA,扫描速率为6°·min-1。采用日本JEOL公司生产的JSM 6460电子扫描电镜测试样品的形貌。采用美国NICOLET公司生产的NEXUS 670傅立叶变换红外光谱仪进行测试。利用固体KBr压片法制样。分辨率:4 cm-1;扫描次数:32次;测试范围 4 000~400 cm-1。
2 结果与讨论
2.1 离子液体的核磁共振光谱
用DMSO作为溶剂对合成的离子液体进行了核磁共振分析,离子液体的结构得到了进一步证实[19]。主要数据见图1。
2.2 羟基磷灰石的傅立叶红外光谱
图2是以不同的离子液体为溶剂所制备的羟基磷灰石的红外谱图。可以看出,图2中4个样品的红外谱图基本一致,与已报道的羟基磷灰石的红外谱图基本相符。这具体表现在以下几个吸收峰上。首先,960~1 100 cm-1为P-O的伸缩振动吸收峰,540~620 cm-1为O-P-O的弯曲振动吸收峰。在1 450,1 400和870 cm-1处分别为CO32-的伸缩振动和弯曲振动吸收峰。这说明有少量的CO32-取代了PO43-。可能是由于所使用的原料氢氧化钙长期暴露在空气中,部分变质为碳酸钙,从而引入了少量的CO32-。630 cm-1处为羟基的弯曲振动吸收峰,3 570 cm-1处为羟基的伸缩振动。另外,在1 650和3 500 cm-1处是表面吸附水分子的羟基振动峰。处于3 150 cm-1处的吸收峰可归属为表面水分子与HAP中的羟基相互作用产生分子间氢键后而引起的羟基振动峰。另外,在2 900 cm-1附近出现弱的C-H弯曲振动峰,也就意味着由于洗涤不够充分,合成的羟基磷灰石内部或者表面有残留的少量离子液体。
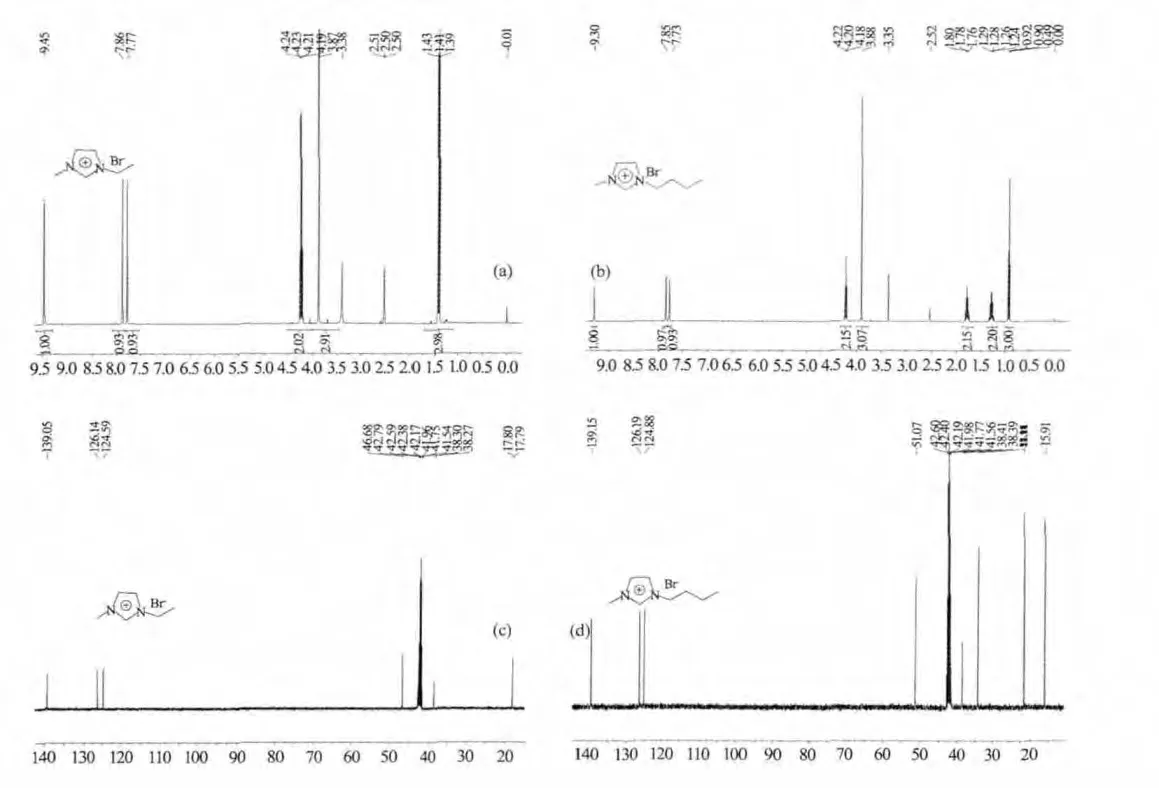
图1 不同的离子液体的核磁共振谱谱图Fig.1 1H NMR spectra of EmimBr(a)and BmimBr(b),and13C NMR spectra of EmimBr(c)and BmimBr(d)

图2 以不同的离子液体为溶剂制备得到的羟基磷灰石的红外谱图Fig.2 FTIR spectra of the as-synthesized hydroxyapatite with different ionic liquids as solvent
2.3 羟基磷灰石的X-射线粉末衍射图
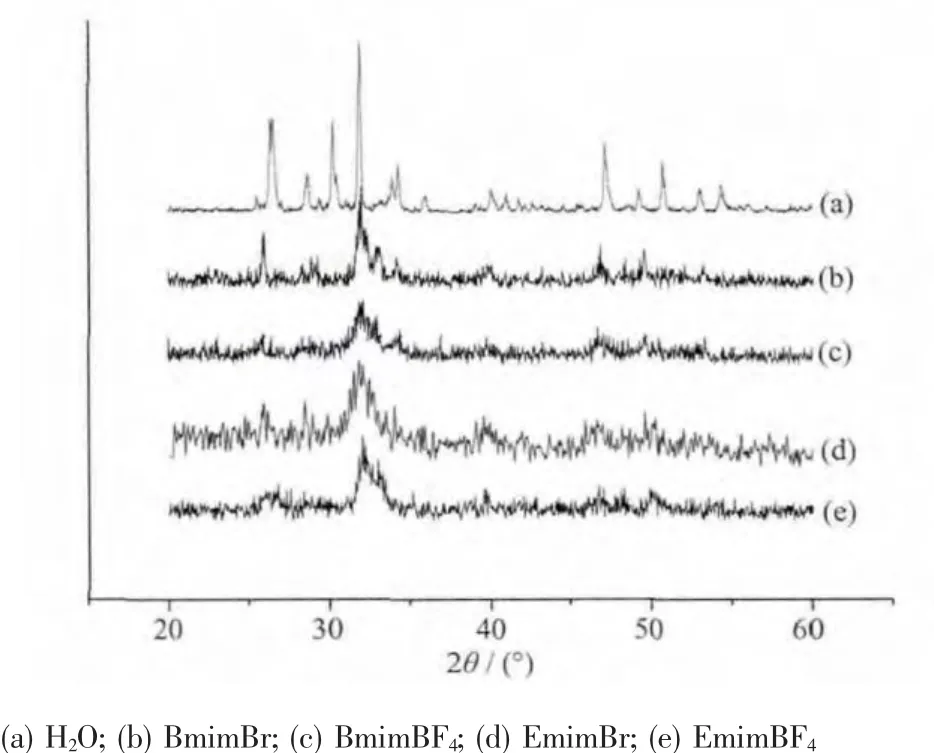
图3 以不同的离子液体为溶剂制备得到的羟基磷灰石的X-射线粉末衍射图Fig.3 XRD patterns of the as-synthesized hydroxyapatite with different ionic liquids as solvent
图3为离子液体中制备的羟基磷灰石的X-射线粉末衍射图。从所得到的衍射图谱中可以看出,其与已报道的羟基磷灰石的衍射峰位置基本一致。与水热法制备的羟基磷灰石不同,离子热合成的羟基磷灰石XRD衍射峰明显不够尖锐,这是由于颗粒较小以及羟基磷灰石晶格中有少量的CO32-取代PO43-,从而导致结晶度降低,但也说明了我们所合成的羟基磷灰石的活性是比较高的。因为,在人体骨组织中,羟基磷灰石晶体是属于弱晶范畴的[20],本文中合成的羟基磷灰石在结晶度上与人体骨中的羟基磷灰石结晶体相类似,这有力的拓宽了其作为生物仿生材料的应用范畴。除此之外,我们可以发现图3给出的四个样品的粉末衍射图基本一致,这说明离子液体在羟基磷灰石合成过程中对其晶体结构的形成基本没有影响。
2.4 羟基磷灰石的扫描电子显微镜
图4为分别以水和4种不同的离子液体为溶剂,所合成的羟基磷灰石的扫描电子显微镜照片。从图中可以看出,制备过程中所使用的介质不同,所得到的羟基磷灰石的形貌不同。以水为溶剂所制备的样品(图4a),其颗粒的均一性不好,团聚现象较为明显,颗粒尺寸从纳米级到2~30 μm不等,而以室温离子液体为溶剂所制备得到的样品 (图4b,c,d,e),其颗粒尺寸及颗粒的形貌均较为规则。这充分说明,离子液体在羟基磷灰石的合成过程中起到了修饰的作用,可以调变其颗粒的尺寸,形貌等。图4b是以离子液体BmimBr为溶剂所合成的羟基磷灰石的SEM照片,从照片中可以观察到珊瑚状的纳米结构。当把离子液体换成BmimBF4时(图4c),出现很多微米球,这些微米球的尺寸基本相同,并且在这些微米球的周围,缠绕着许多的纳米纤维。比较图4b和图4c,便可以发现所使用的离子液体的阳离子(Bmim+)不变,阴离子由Br-变为BF4-时,所合成样品颗粒的尺寸,形貌更加规则,均一。当溶剂使用EmimBr时(图4d),从图中可以看到一些球状的聚集体,尺寸约为0.5~4 μm。从图 4e中可以观察到一些小的球状的颗粒上长出一些宽约150 nm,长约1 μm的纳米棒。比较图4c和图4e,可以发现,当所使用的离子液体的阴离子(BF4-)不变,阳离子由Bmim+变为Emim+时,所制备的颗粒的尺寸及形貌更加规则。通过5个样品的SEM照片可以得出结论,在所使用的离子液体中,EmimBF4更有利于合成尺寸较小,形貌规则的羟基磷灰石粒子。由于离子液体的阴阳离子的极化率不同,将会影响它自身聚合的趋势,从而起到表面活性剂的作用[21]。我们都知道,通过调变离子液体的阴阳离子的结构,可以调变它的极化率,粘度等溶剂性质。而在材料合成过程中,包括溶剂性质在内的这些物理化学性质起到非常重要的作用。因此,我们通过改变阴离子和阳离子碳链的长度来改变离子液体的极化率,从而实现了对羟基磷灰石超细粉体形貌的

图4 不同溶剂制备得到的羟基磷灰石的扫描电子显微镜照片Fig.4 SEM micrographs of the as-synthesized hydroxyapatite with different ionic liquids as solvent
调变。另外,本文所采用的离子液体亲水性较强而导致容易吸水,因此在离子热合成过程中离子液体的吸水量多少对形貌的影响也值得我们做进一步深入的研究。
3 羟基磷灰石超细粉体在染料吸附中的应用
由于羟基磷灰石对生物大分子具有良好的吸附性,这些年来,人们对其吸附性能也做了大量的研究。人们己经研究了羟基磷灰石对短链有机物、高分子聚合物[22]、唾液蛋白[23]、接触霉素[24]、溶解霉素[25]、蛋白质[26-27]的吸附。另外,羟基磷灰石(HAP)由于其特殊的晶体化学特征,使得它对多种金属阳离子具有广泛的容纳性和吸附固定作用,从而成为一种新型的环境功能矿物材料。例如,羟基磷灰石对水溶液中的F-和Pb2+有很好的交换性能和去除作用,因而成为专用无机吸附材料。本文中,我们分别以在H2O和BmimBr中合成的羟基磷灰石为例,考察了其在水体系中对有机染料碱性品红的吸附性能。
以H2O和BmimBr为溶剂所合成的羟基磷灰石材料在水溶液中对碱性品红的吸附量随时间的变化曲线在图5中给出,在初始浓度为100 mg·L-1的碱性品红溶液中(10 mL)分别加入10 mg的羟基磷灰石材料。可以看出,在吸附300 min,吸附达到平衡时,它们在此条件下的饱和吸附量分别为43.78和30.15 mg·g-1。在达平衡后,以离子液体为溶剂所合成的羟基磷灰石材料对碱性品红的吸附量更大。但由于离子热合成的羟基磷灰石表面或内部有残留的少量离子液体,对碱性品红的吸附产生了负面影响,导致吸附效果并没有明显增大。总之,吸附实验显示了以离子液体为溶剂所合成的羟基磷灰石材料较为优良的吸附性能。这与离子液体在合成过程的修饰作用有关,使得离子液体中所合成的羟基磷灰石材料的颗粒规则,尺寸较小,比表面积较大,从而使其能更加有效的吸附碱性品红染料分子。
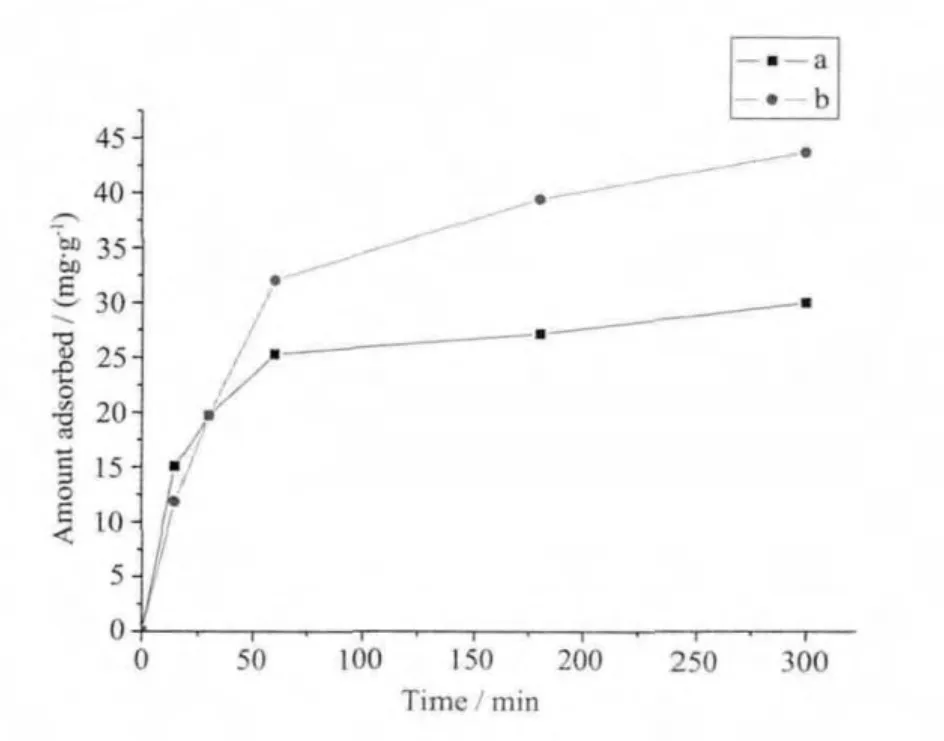
图5 以H2O(a)和BmimBr(b)为溶剂所合成的羟基磷灰石材料对碱性品红的吸附动力学曲线对照图Fig.5 Adsorption kinetics of organic dyes rosaniline on as-synthesized hydroxyapatite with(a)H2O and(b)BmimBr as solvent
4 结 论
我们以室温离子液体作为溶剂,以氢氧化钙为钙源,磷酸为磷源,提出了一种新的无需高温焙烧制备羟基磷灰石超细粉体的方法。所得到的羟基磷灰石为部分CO32-取代的羟基磷灰石(原料中氢氧化钙部分变质为碳酸钙,引入了少量的CO32-),其组成接近人体骨组织中的羟基磷灰石组成。离子液体在合成过程中起到了修饰剂,分散剂的作用。我们通过调变离子液体的阴阳离子,实现了对羟基磷灰石超细粉体形貌的调变,为进一步实现对其形貌的调控奠定了基础。另外,此合成方法利用离子液体蒸汽压低,不易挥发,可重复利用等优点降低了合成成本。以离子液体BmimBr作为溶剂所合成的羟基磷灰石材料对有机染料碱性品红具有较好的吸附性能。
[1]Hideki A.Medical Application of Hydroxyapatite.Tokyo:Ishiyaku Euroamerica Inc,1994:90
[2]Kano S,Yamazaki A,Otsuka R,et al.Bio-Med.Mater.Eng.,1994,4(4):238-290
[3]Brennecke J G,Maginn E J.J.AICHE,2001,47(11):2384-2389
[4]Seddon K R.J.Chem.Technol.Biotechnol.,1997,68(4):351-356
[5]Wasserscheid P,Keim W.Angew.Chem.Int.Ed.,2000,39(21):3772-3789
[6]Fujii S,Akiba U,Fujihira M.Chem.Commun.,2001,17:1688-1689
[7]Shelton R.Chem.Commun.,2001,23:2399-2407
[8]Wheeler C,West K N,Liotta C L,et al.Chem.Commun.,2001,10:887-888
[9]Olivier-Bourbigou H,Magna L.J.Mol.Catal.A:Chem.,2002,182-183:419-437
[10]Huddleston J G,Willauer H D,Swatloski R P,et al.Chem.Commun.,1998,16:1765-1766
[11]FadeevAG,MeagherMM.Chem.Commun.,2001,3:295-296
[12]Lo W H,Yang H Y,Wei G T.Green Chem.,2003,5:639-642
[13]Zhang S G,Zhang Q L,Zhang Z C.Ind.Eng.Chem.Res.,2004,43(2):614-622
[14]Cooper E R,Andrews C D,Wheatley P S,et al.Nature,2004,430(7003):1012-1016
[15]Hideaki I,Kensuke N,Yoshiki C.J.Am.Chem.Soc.,2004,126(10):3026-3027
[16]Kim K S,Demberelnyamba D,Lee H.Langmuir,2004,20(3):556-560
[17]Suarez P A Z,Dullius J E L,Einloft S,et al.Polyhedron,1996,15(7):1217-1219
[18]Holbrey J D,Seddon K R.J.Chem.Soc.,Dalton Trans.,1999,13:2133-2140
[19](a)ZHANG Wen-Xiu(张文秀),CHEN Yong-Jie(陈永杰),LI Xing(李 行),HUANG Zhi(黄 志).Journal of Shengyang University of Chemical Technology(沈阳化工大学学报),2012,26(1):9-12(b)Huang P,Zhai M L,Peng J,et al.Carbohydr.Polym.,2008,71:690-693
[20]Suchanek W L,Shuk P,Byrappa K,et al.Biomaterials,2002,23(3):699-710
[21]Antonietti M,Kuang D,Smarsly B,et al.Angew.Chem.Int.Ed.2004,43(38):4988-4992
[22]Tsortos A,Nancollas G H.J.Colloid Interface Sci.,1999,209(1):109-115
[23]Kawasaki K,Kambara M,Matsumura H,et al.Colloids Surf.B:Biointerfaces,2003,32(4):321-334
[24]Barroug A,Lemoux E,Lemaitre J,et al.J.Colloid Interface Sci.,1998,208(1):147-152
[25]Aizawa T,Oganesawa N,Kamakura A,et al.FEBS Lett.,1998,422(2):175-178
[26]Luo Q,Andrade J D.J.Colloid Interface Sci.,1998,200(1):104-113
[27]Wassell D T H,Hall R C,Embery G,et al.Biomaterials,1995,16(9):697-702
