溶胶-凝胶过程中有机硅氧烷的自组装行为研究进展
2014-07-02宋少飞胡道道沈淑坤李伟
宋少飞,胡道道,沈淑坤,李伟
(1陕西师范大学材料科学与工程学院,陕西 西安 710062;2运城学院应用化学系,山西 运城 044000)
溶胶-凝胶过程中有机硅氧烷的自组装行为研究进展
宋少飞1,2,胡道道1,沈淑坤1,李伟1
(1陕西师范大学材料科学与工程学院,陕西 西安 710062;2运城学院应用化学系,山西 运城 044000)
在有机硅氧烷的溶胶-凝胶过程中,常常形成超分子自组装结构,这种自组装行为对前体水解/缩合反应过程具有非常重要的影响。本文从有机硅氧烷分子自身结构影响和外源分子的诱导作用两个方面出发,综述了近年来有关溶胶-凝胶过程中有机硅氧烷自组装行为研究方面的特色工作。分析了该领域未来发展的主要方向,指出设计合成含有独特官能团的有机硅氧烷为前体,引入外源分子,基于外源分子与前体分子之间的相互作用构筑有机硅氧烷超分子体系,利用水解/缩合过程与自组装体系之间的协同作用制备具有长程有序的硅基复合材料,将是该领域未来研究的重点。
有机硅氧烷;水解;自组装;二氧化硅;复合材料
在过去的二十几年里,硅基复合材料由于其独特的结构和性质,受到人们的广泛关注。硅基复合材料兼具有机物与无机物的特性,材料中无机组分Si—O—Si网络为复合材料提供了基本骨架,赋予了其较为优异的热稳定性和刚性等特性;而材料中的有机成分,基于其内在的可塑性、柔韧性以及具有化学活性官能团的功能化等特性,能够有效调节硅基复合材料的物理性能,且有利于材料的进一步修饰,赋予其许多新颖的性质与功能[1]。一系列具有不同结构特征的有机硅氧烷、聚倍半硅氧烷材料、有序介孔硅基材料、功能薄膜等复合材料被相继制备[2-3]。
在有关硅基材料的制备研究中发现,当使用有机修饰的硅氧烷为前体时,与小分子硅氧烷(如TEOS或TMOS)不同,由于前体分子中Si原子上取代基之间的非共价键作用(氢键、范德华力、π-π堆积作用和静电作用等),使前体或其水解产物在反应体系中表现出超分子自组装行为,而且这种自组装行为对前体水解/缩合行为具有非常重要的影响。为此,从分子尺度理解和控制这种自组装行为的相关研究得到广泛重视。研究表明,溶胶-凝胶过程中有机硅氧烷的自组装行为受到前体中烷基结构、外源引入分子和实验条件等诸多因素的影响[4-5]。如Loy等[6]研究了含不同有机取代基的有机硅氧烷前体在溶胶-凝胶过程中的簇集行为。实验发现,含甲基、乙烯基、氯甲基等小分子取代基的三烷氧基硅氧烷,易于水解/缩合形成透明的块体凝胶,含有长链取代基(十二烷基、十八烷基)的前体也容易形成凝胶。作者认为,后者之所以能形成凝胶,与长链烷基间的自组装作用有关。在本实验室早期的工作中[7],曾借助荧光探针技术在线跟踪了前体甲基丙烯酰氧丙基三甲氧基硅烷(MAPTMS)在本体相中的水解/缩聚过程,基于所观察到的荧光探针的荧光光谱涨落行为,首次提出了这一前体的水解产物具有不同于小分子前体的两亲性,前体水解产物的胶束簇集与解簇集是导致荧光探针光谱涨落的主要原因。另外,Inagaki等[8]在碱性溶液中,使用联苯基桥联的有机硅氧烷[(C2H5O)3Si-(C6H4)2-Si(OC2H5)3]为前体,十八烷基三甲基氯化铵为表面活性剂,成功制备了一种联苯基桥联的有序介孔倍半硅氧烷材料。该介孔材料的孔壁具有层状和中尺度的有序性结构。这种晶型孔壁的形成是由于层状孔壁中的联苯基桥联前体通过疏水亲水相互作用力进行自组装导致的。
上述几例研究结果均表明,以具有自组装功能的有机硅氧烷为前体,或者在溶胶-凝胶体系中加入表面活性剂分子,皆可以在水解/缩合过程中构筑超分子体系。溶胶-凝胶过程中有机硅氧烷的自组装行为对反应动力学及热力学性质、组织排布方式、空间结构演化、物种存在状态、产物微区环境、产物结构等均具有非常重要的影响。值得说明的是,众所周知,超分子体系中的构筑单元一般为非反应性,使其自组装形成的高度有序集合体具有静态特征。而对于有机硅氧烷溶胶-凝胶过程中的超分子自组装体系而言,因前体硅氧烷基团在自组装过程中的不断反应,使簇集行为与有机硅氧烷水解/缩合作用动态过程密切相关,从而使这些体系中的超分子作用更为复杂,导致前体水解/缩合行为更为复杂。因此,有机硅氧烷前体在溶胶-凝胶过程中自组装行为更具特殊性。同时,也正是簇集行为与水解/缩合反应的关联性,为控制性制备有机无机复合材料带来挑战与机遇。下文主要从前体结构影响和外源分子诱导作用两个方面出发,对有机硅氧烷水解/缩合过程中的自组装行为进行简要综述。
1 前体自身结构导致的自组装
尽管有机硅氧烷种类繁多,但依据其Si原子上烷基取代基结构特征,可将其分为短链有机硅氧烷、长链有机硅氧烷、桥联型有机硅氧烷以及树形有机硅氧烷等[9-11]。有机硅氧烷分子中取代基这种结构特点以及其性质差异(空间位阻效应、电子效应及官能团化学反应活性等)必然对其在体系中的分散行为产生影响。事实上,并非所有有机硅氧烷水解/缩合体系均存在自组装行为,而是含有某些特征官能团的前体分子能够在其溶胶-凝胶过程中形成超分子自组装体系。以下按照有机硅氧烷前体自身结构特征,简述其水解/缩合体系中的典型自组装行为。
1.1 类表面活性剂型前体
含有疏水性有机基团的硅氧烷,在水溶液中,其水解产物由于无机端Si—OH更好的亲水性,具有类似表面活性剂的结构特点,从而使其具有双亲性。对于含有较长疏水基的前体,这一特性表现得更为突出,也使其水解/缩合产物自组装行为更为明显。
例如,基于含长链烷基前体水解产物的双亲性,在固体表面取向规整、排列紧密的自组装单分子膜被成功制备[12]。Allara等[13]研究结果表明,基质表面预水化对制备高度有序的单分子膜非常必要。当膜与基质界面间存在超薄的物理吸附水层时,所形成的单分子膜具有最高结构有序度。这种高的有序度与长链硅氧烷水解产物在准二维方向上的自组装行为密切相关。水解初期,利用烷基链间的范德华力,水解产物分子进行自组装,侧向移动而相互靠近在膜-水界面形成覆盖面较小的Langmiur膜。随着单分子膜覆盖面积增加,水解产物中的硅醇之间发生缩合反应,形成Si—O—Si键而交联,最终形成有序单分子膜。
基质界面上二维溶胶-凝胶过程的研究有助于前体水解/缩聚反应机制的深入理解及实验条件的选择。如Leblanc等[14]利用Langmuir-Blodgett技术,并结合Brewster角显微镜表征手段,在pH值为1.4~12.8范围内,研究了十八烷基硅氧烷(C18TMS)在气/液界面自组装成Langmiur膜的过程。研究发现,无论是在酸性底相还是碱性底相中,前体水解/缩合均形成线性结构高聚物。该作者认为,十八烷基链之间的空间位阻效应和疏水作用是实现自组装的驱动力,通过自组装作用缩聚产物形成定向有序排列结构,疏水基团均匀分布在缩聚链的两侧。
类表面活性剂前体除了可以在界面上自组装形成二维结构的有序薄膜外,在本体相中也将形成高度有序的三维超分子自组装体系。为了验证这一想法,Parikh等[15]在水中进行了十八烷基三氯硅烷的水解/缩合实验,得到了一种层状微晶高聚物(聚十八烷基三氯硅烷,结构示意见图1),证实了上述预想的可行性。从微观结构分析,硅氧烷水解产物具有双亲性,在水中通过自组装形成头对头双层堆积。在这里,高度有序层状高聚物形成的关键是水解产物自组装形成硅醇双层堆积结构,然后硅羟基在同一平面进行缩合反应,即先水解,再自组装,最后缩合交联。另外,长链烷基前体水解过程中的自组装行为也被成功用于多层有序聚集体的自发合成。Kuroda等[16]通过长链硅氧烷与TMOS混合前体的水解/缩合反应,利用旋涂法成功制备了有序透明杂化薄膜。杂化薄膜的形成被归因于长链硅氧烷的自组装行为与TMOS在溶胶-凝胶过程中可以形成Si—O—Si网络能力的共同作用。在该小组随后的工作中[17],他们进一步研究了烷氧基数目和有机链长度[CnH2n+1(CH3)mSi(OMe)3-m,m=0~2,n=8~12]对杂化薄膜形貌和结构的影响。结果表明,除了三甲氧基硅氧烷外,单甲氧基和双甲氧基硅氧烷前体也可以与TMOS通过共缩合形成有序多层薄膜,但随着Si原子上烷氧基数量的不同,层间距d也发生变化。他们认为在共混前体水解/缩合过程中,生成了既含有长链烷基又带有硅醇的两亲性硅氧烷低聚物,正是由于低聚物的自组装行为导致了多层有序薄膜材料的形成。多层薄膜是由共价链接到硅氧单层的烷基双层排列组成的。通过这种方法,可以实现外观均一、结构有序以及纳米层定向排列的纳米杂化薄膜的可控性制备。
此外,Chemtob等[18-19]研究了长链烷基硅氧烷十二烷基三甲氧基硅烷溶胶-凝胶过程中的自组装行为,考察了紫外光照、反应温度及体系pH值等对该过程反应动力学及热力学的影响。实验结果表明,长链有机硅氧烷前体在溶胶-凝胶过程中的自组装行为是由于烷基链之间的范德华力及疏水作用造成的,产物结构经历了一个从无序到有序的转化过程。Fan等[20]利用双亲性长链硅烷前体十八烷基二甲基(3-三甲氧基硅丙基)氯化铵的自组装行为,通过溶胶-凝胶过程,成功实现了对疏水性纳米金的表面功能化。纳米金表面烷基链与前体长烷基链之间的疏水作用是实现包裹的驱动力。包裹后的纳米金粒子表面由疏水变为亲水,在水溶液中单分散性好,而且可以在体系中形成空间三维有序排列。
除了长链烷基以外,前体分子中烷基链上带有某些可质子化的官能团时,对其在体系中的簇集行为也会产生重要影响。本实验室[21]以Pyranine为荧光探针,利用静态荧光光谱在线跟踪了烷基链上带不同官能团的有机硅氧烷水解/缩合反应过程。结果表明,在酸性条件下,当分子中含有短链有机基团或者含有可质子化环氧基时,前体的水解产物没有明显的双亲性,均匀分散于水相。而对于含酯基的长链前体,由于酯基的疏水性,其水解产物具有类似表面活性剂分子的双亲性,在水相中通过分子自组装形成胶束结构,导致体系成为存在疏水微区的不均相体系。同时,从荧光探针在线检测结果发现,由于前体取代基的不同,所导致的水解产物在溶液中的存在方式差异会显著影响水解/缩合反应的动力学过程,从而影响最终缩合产物的物种形态。其中,含强疏水性基团前体的缩合产物易于发生凝聚而从体系中析出。
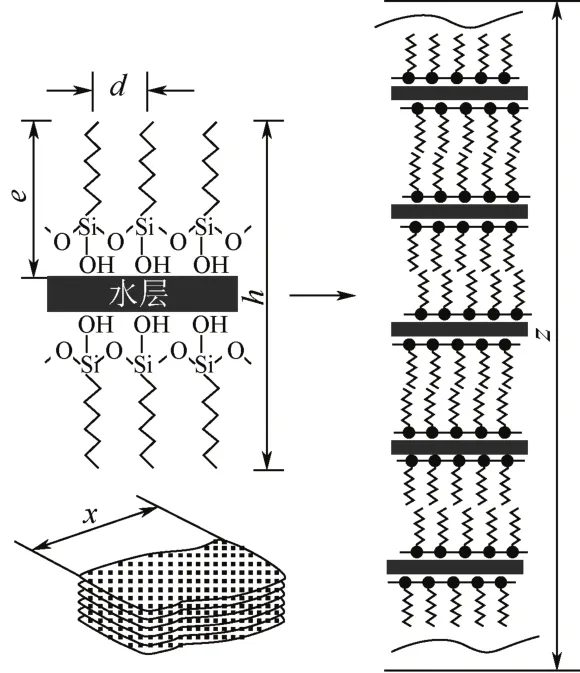
图1 PODS的结构示意图
1.2 Bola型前体
在众多的有机硅氧烷分子中,桥联型有机硅氧烷具有与Bola表面活性剂相似的结构特征,即中间为疏水链,两头各含一个极性基团的典型结构。在气液界面和水中簇集行为丰富,可以通过自身分子的自组装形成超分子体系,近年来受到人们的普遍关注[22]。桥联型硅氧烷分子中,作为桥联基团的有机组分可以是柔性烷基链,也可以是含有不饱和结构的刚性链,也可以根据实际需要对桥联基团链长、共价结合位点以及功能性等方面进行调整,从而实现对材料的理化性质进行精细调控。如聚合的桥联硅氧烷依据引入的有机骨架的长短尺度和刚性差异而呈现不同的三维网络结构。如果连接臂是较短的烷基链,得到的聚合物具有多孔结构;连接臂较长,则柔性增加,孔道结构容易塌陷。但是当引入刚性的连接臂时,如有机链中含有苯环或烯、炔键,则会导致材料多孔性。利用桥联型硅氧烷为前体制备的桥联聚倍半硅氧烷复合材料在材料组成上易于调控,被广泛应用于表面改性剂、涂料、催化剂和膜材料等领域[23]。
当桥联型硅氧烷分子中含有苯基、炔基、联苯基及其衍生物等刚性连接臂时,在前体的溶胶-凝胶过程中常常会出现超分子自组装行为。Corriu等[24]使用1,3,5-三(对三甲氧基硅基苯基)苯和1,3,5-三(三甲氧基硅基丙基苯基)苯为前体进行了水解/缩合反应,制备了纳米复合材料,并使用双折射检测与XRD技术对材料进行了表征。研究结果表明,刚性片层核周围柔性链的存在对于所制备材料的形貌及双折射性具有非常大的影响。柔性链的存在更有利于分子各向异性的自组装行为,在水解/缩合过程中,相关物种利用连接臂之间的疏水作用、范德华力、π-π堆积等弱相互作用力进行了自组装,调节了材料结构的有序程度,所制备复合材料具有较好的各向异性特征。Creff等[25]研究了双脲基苯基桥联的有机硅氧烷在溶胶-凝胶过程中的自组装行为,水解/缩合反应结束后,得到一种针状单晶纳米材料。基于相关实验表征数据,作者认为,在溶胶-凝胶反应前期,前体分子利用桥联基分子间的非共价作用形成了自组装体系,有利于其前期的水解。反应后期,通过前体分子中硅氧烷基之间缩合形成的Si—O—Si共价键与桥联基分子间的氢键协同作用形成高度有序复合材料。最近,中国科学院化学所Fu等[26]设计合成了一种苝酰亚胺基桥联型硅氧烷前体,通过前体水解产物硅醇分子间的氢键作用和π-π堆积作用形成自组装体,接着与三甲基氯硅烷进行共聚缩合,制备了一种结构完美的梯形聚倍半硅氧烷材料。该材料保持了苝酰亚胺基团良好的光电性质,可作为电子接受体用于聚合物太阳能电池方面。另外,该聚合材料在有机溶剂中具有较好的可溶性和较高的热稳定性。
对于由共轭聚合物形成的电子器件来讲,很难实现材料微米级的高有序单失畴(指液晶高分子的分子链有很窄或单一取向的有序区域)结构。为了解决这一难题,从分子水平上调控生色团的分子聚集行为,Dautel等[27]以二苯乙烯基苯酰胺生色团为桥联基合成了一种桥联型有机硅氧烷,通过旋涂制膜法分别制备了其单体溶液与水解/缩合产物溶液的薄膜,对比了两种薄膜中生色团的排列方式,探讨了溶胶-凝胶过程中物种簇集行为对分子中生色团簇集行为的影响。实验结果表明,单体在玻璃片上形成的沉积物中,由于分子的全反式构象和Si—(OC2H5)3基团的空间位阻效应,生色团自组装形成头对尾排列的J型聚集体。而其水解/缩合产物所形成的薄膜中,由于Si—(OC2H5)3基团水解/缩合所导致的侧位滑移,生色团则形成面对面堆积的三明治结构的H型聚集体。这两种生色团不同的聚集行为赋予两种材料完全不同的光电性质。
1.3 含特殊官能团取代基前体
为了调控材料的结构和性质,赋予其新的功能,研究人员别具匠心地设计了很多具有自组装能力的含特殊官能团连接臂的有机硅氧烷、特殊官能团间的相互作用,赋予了这类有机硅氧烷独特的自组装行为。例如,一种含有手性结构酰胺功能基团的硅氧烷被设计合成[28],以其为前体,通过溶胶-凝胶过程,一种具有手性形貌的复合材料被首次制备,该材料具有双螺旋结构。作者认为,类似于DNA分子的结构,氢键作用是主要的自组装驱动力。水解/缩合过程中,前体水解产物或缩合的低聚物之间形成氢键弱相互作用,诱导Si—O—Si键的定向生成。这类具有手性形貌和性质的复合材料在非均相手性催化领域有重要的潜在应用价值。以类似驱动力的有机硅氧烷为前体,可以合成具有有序结构的功能化材料。如图2所示,通过前体分子中有机基团之间的氢键作用,制备得到含大环冠醚的有序结构复合材料[29]。显然,这种材料可通过冠醚实现对碱金属及碱土金属离子的选择性结合。

图2 含有大环冠醚受体的杂化材料结构示意
另外,高分子化合物也被引入有机硅氧烷分子中,用于调控自组装行为。Velamakanni等[30]合成了一种三嵌段硅基聚合物,研究了该聚合物在不同界面上的自组装行为。结果表明,通过硅氧烷基与基质表面羟基的水解/缩合作用,该聚合物可以在基质表面进行自组装,形成有序的单层结构。而且所形成的表面薄膜结构完整,具有非常好的稳定性和耐热、耐强酸强碱腐蚀性。尾端带有二茂铁基团的有机硅氧烷被用于基质的表面修饰,通过二茂铁基团之间的相互作用在基质表面形成有序单层[31]。
2 外源分子诱导的自组装
相对于前体分子自身结构导致的自组装而言,通过向体系中引入外源分子所形成的超分子自组装行为作用方式更为简单灵活,产物组成及结构更加丰富多样,其对前体溶胶-凝胶过程的影响也更加复杂。一般而言,外源分子主要通过“模板作用”,利用自身的自组装行为对前体簇集行为进行调节,在溶胶-凝胶过程中形成自组装体系,从而对前体水解/缩合行为造成重要影响。这种影响体现在外源分子结构对杂化材料微观形貌、尺寸和结构等方面的调控。
2.1 表面活性剂分子
在硅基杂化材料的溶胶-凝胶法制备工艺中,表面活性剂分子常被引入到体系中,用于调控材料的结构和性质。陈连喜等[32]在碱性条件下,以表面活性剂十二烷基苯磺酸钠为模板,乙烯基三乙氧基硅烷为前体,通过溶胶-凝胶法成功合成了不同粒径、高度单分散的纳米级球形粒子。Landskron等[33]使用环状结构的三环倍半硅氧烷[(EtO)2Si(CH2)]3为前体,利用表面活性剂的模板作用,通过前体与表面活性剂的自组装行为合成了内部含有交联结构的[Si(CH2)]3环的杂化有序介孔薄膜。分析结果表明,该材料介电常数较低,而且具有较高的热稳定性,在微电子领域具有潜在的应用价值。
桥联型倍半硅氧烷介孔材料是一类新型功能材料,它是用桥联型有机硅氧烷作为前体,表面活性剂为模板,通过溶胶-凝胶过程,利用模板分子间弱相互作用力进行自组装形成的具有规则孔道结构的杂化多孔功能材料。如Inagaki等[34]以苯基桥联的有机硅氧烷为前体,十八烷基三甲基溴化铵为表面活性剂,成功制备了具有六边形介孔结构并且结晶状孔壁的有序介孔硅杂化材料。这种材料的有序结构被归因于前体之间以及前体与表面活性剂分子之间的相互作用所导致的自组装行为,其结晶状孔壁的形成是由于前体分子中苯基与硅氧基亲疏水性交替排列的结果。最近,利用十六烷基三乙氧基溴化铵为模板,HCl为催化剂,通过TEOS与乙基桥联的有机硅氧烷1,2-二(三乙氧硅基)乙烷的共缩聚反应,Pan等[35]成功制备了具有周期立方相的介孔硅基杂化材料,所得到的乙基桥联的杂化材料具有很高的水热稳定性。
具有手性结构的表面活性剂分子由于其较强的立体结构特征,被用于手性特征杂化材料的制备。例如,Marx等[36]使用手性阳离子表面活性剂(-)-N-十二基-N-甲基麻黄碱溴化物为模板,TEOS与苯基三甲氧基硅烷为共聚前体,制备了一种对立体异构体具有选择性识别能力的分子印迹杂化材料,并对其识别机理进行了探讨。他们认为,模板分子在溶胶-凝胶过程中的自组装能力对于材料的识别功能具有重要的决定作用。
一些结构特殊的、含活性官能团的表面活性剂也被用于调控前体水解/缩合过程中的自组装行为。例如,Pinnavaia等[37]使用Bola型双亲表面活性剂NH2(CH2)nNH2为结构导向剂,通过与前体TEOS自组装制备了具有层状结构的分子筛。研究发现,随着导向剂分子中烷基链的长度增加,分子筛的孔径逐渐变大,而且呈现不同的形貌。作者提出了如图3所示自组装材料的形成机理。TEOS的疏水性导致溶液中产生疏水-亲水相分离和囊泡的产生,同时产生水/油界面,部分水解的前体产物通过和氨基的氢键作用也包含在水层,由此囊泡包含一层层被水和硅醇分开的表面活性剂。然后,在这一自组装体基础上进行缩合反应,导致了二氧化硅颗粒的层状堆积。Kim等[38]研究发现少量酸性表面活性剂全氟代十四烷酸(PFTDA)可加速气/液界面上十八烷基三甲氧基硅烷(ODTMS)单分子膜的水解/缩合速度,有利于水解产物的自组装行为,促进凝胶形成。由于PFTDA较强的酸性,在体系中集中于硅氧烷甲氧基基团附近,导致水解反应局部酸度较高,加快了ODTMS的水解速度,提高了前体水解程度。同时,水解产物硅醇由于Si—OH数目的增加,具有更强的自组装能力。上述研究结果表明,通过加入PFTDA可以实现调节膜层的水解/缩合速度及水解产物自组装行为。这种调节与控制作用对于实现二维聚硅氧烷材料在催化、分离、传感等方面的应用具有重要的指导意义。
使用混合表面活性剂为模板,与单独使用其中一种表面活性剂模板所导致的前体自组装行为完全不同,通过混合表面活性剂分子之间的相互作用,可以制备一些结构独特的功能材料。例如,Wang等[39]利用10,12-二炔基-二十五烷酸(PDA)和它的衍生物N-(11-O-α-D-吡喃葡糖-3,6,9-三氧杂)十一烷基10,12-二炔基-二十五烷酸(PDTMC)与十八烷基三甲氧基硅烷(ODTMS)在界面上进行化学及光化学双聚合反应,制备了Langmuir单分子膜,膜结构如图4所示。研究表明,通过调节底相溶液的pH值,PDA/PDTMC的光聚合反应和ODTMS的缩合反应能够相互影响。在该自组装体系中存在两种驱动力,即前体ODTMS分子中较长碳链之间的疏水作用,水解产物硅醇和PDA分子中羟基以及PDTM分子中酰氨基之间存在的氢键作用。在适当pH值条件下,混合物分子之间通过自组装形成间隔有序结构,彼此互溶。利用彼此之间的立体位阻效应,导致对方的反应减缓,经过共聚形成自组装单层。Meng等[40]使用非手性的阳离子氟化表面活性剂[CF3(CF2)3SO2NH(CH2)3N+(CH3)3I-]和CTAB 为混合模板,乙基或苯基桥联的有机硅氧烷为硅源,通过溶胶-凝胶法,成功制得扭曲和螺旋形六角棒形貌的手性有序介孔有机硅杂化材料。形成这种螺旋结构的驱动力是混合表面活性剂协同作用的结果。

图3 囊泡自组装体系形成机理示意
2.2 其他外源分子
除了常规表面活性剂分子外,一些有机物或高分子化合物也被用于调节有机硅氧烷在溶胶-凝胶过程中的自组装行为,以制备新型功能材料。例如,Silva等[41]使用生物高聚物壳聚糖和硅烷偶联剂3-异氰酸丙基三乙氧基硅烷,通过溶胶-凝胶工艺,制备了一种新型杂化材料。杂化材料中,壳聚糖通过脲基共价链接到硅烷偶联剂的聚硅氧烷链上。研究表明,该材料具有有趣的光致发光特性和生物活性。Ariga等[42]以一种类似“蜥蜴”型的分子作为模板,设计制备了具有特异结构和功能的介孔二氧化硅材料。该模板分子具有可以与硅氧烷基团进行共价缩合的阳离子头基和疏水性的烷基链尾部,分子中间为欲赋予材料特性的功能基团,模板分子的疏水尾可以赋予前体自组装性质。
本文作者课题组[43]研究发现,N-苯基甘氨酸(NPG)能够改变甲基丙烯酰氧基丙基三甲氧基硅烷(MAPTMS)前体的自组装行为,加速体系的胶凝,并在此基础上提出了NPG协助MAPTMS自组装的机理。MAPTMS 水解产物具有两亲性而易于通过自组装形成胶束,具有一定双亲性的NPG 分子通过“穿插”与水解产物形成共簇集体。这种共簇集体中前体水解产物被插入分子NPG隔离,阻碍了硅醇间的缩合,从而使前体硅氧烷水解更完全,形成具有潜在强交联作用的多羟基单体。随着水解产物单体的增加,体系中形成具有类似结构的高浓度微乳液胶体。随着水解反应进行,胶束中水解产物甲醇浓度的提高以及水量减少,使胶束结构难以继续维持,迫使分子重新排列。此时,具有强缩合能力的前体硅醇解脱了NPG隔离而迅速缩合,从而使MAPTMS 水解体系快速胶凝。人们发现,仅MAPTMS水解/缩合体系很难形成凝胶,因此,上述过程充分体现了分子自组装对有机硅氧烷水解/缩合行为的影响。

图4 PDA/PDTMC/C18TMS形成的单分子膜结构
3 实验条件对自组装行为的影响
超分子自组装不仅与组装单元分子结构有关,也与其所处环境有关,水解/缩合反应条件对于溶胶-凝胶过程中前体自组装行为也具有非常重要的影响,主要表现在对水解/缩合反应速度、物种的簇集方式、溶胶粒子的生长尺度和方向、产物结构与外在形貌的影响等方面。
在硅基材料制备过程中发现,当使用非离子表面活性剂为模板时,一些无机盐的加入会影响产物结构有序性。这种结构上的变化,主要是由于无机盐的加入改变了表面活性剂分子内基团的亲疏水性,从而影响了模板分子自组装过程。例如,Zhai等[44-45]以共聚表面活性剂为模板,在不添加酸的情况下,通过NaCl或FeCl3的协助,成功合成了高度有序的乙基和苯基桥联的介孔有序硅基材料。值得指出的是,在苯基桥联的材料中观察到了部分结晶状的孔壁,当没有加入无机盐时,不会出现这种现象。结合已报道的工作,作者认为无机盐的“盐协助”和“自致生酸”效应是增强前体自组装行为的主要因素。
硅氧烷水解反应是依据SN2亲核取代反应机理进行的,其速控步骤就是亲核试剂水对Si原子的亲核进攻,因此体系中水含量会对前体的水解速率和程度产生影响,导致水解产物的种类和生成次序的变化,从而影响水解/缩合过程中物种的簇集行为。在本文作者已报道的工作中[7],考察了水/硅比对前体水解/缩合行为的影响。研究结果表明,在不同的水/硅比条件下,前体水解/缩合行为有很大差异。低水/硅比时水解速度较快,水解产物具有两亲性,随着反应的进行,体系中不断重复自组装-解组装-自组装的胶束行为。而在水量较多时,水解和缩合速度都被加快,形成了低聚物,破坏了体系的自组装行为。
对于离子型表面活性剂参与的自组装,体系的酸碱性具有重要的影响,甚至影响表面活性剂与前体之间的相互作用,对产物结构有序性产生重要影响。如Markowitz等[46]通过乙基桥联的倍半硅氧烷和N-(2-氨乙基)-3-氨丙基三甲氧基硅烷分别在酸性或碱性条件下共聚合成了有序介孔硅材料。实验表明,在可控制的酸性范围内,可以合成高度有序和较大孔的有序介孔硅材料。
已有研究表明,温度对于分子自组装行为也具有重要的影响[47-48]。Chabal等[49]考察了温度对APTES在二氧化硅基质表面自组装行为的影响。分别在不同温度下通过APTES的水解/缩合反应在二氧化硅基质表面制备了薄膜,使用椭圆光度法和红外光谱研究了薄膜的厚度和组成,比较了溶液温度及后煺火处理对薄膜质量的影响。结果表明,随着溶液反应温度的升高,前体水解/缩合程度越大,所制备的薄膜具有更高的密度和更好的有序结构,70℃时所制备的薄膜密度高、有序性强,而且在水中最稳定。对比实验表明,后退火处理对薄膜结构和性质几乎没有影响。这种温度对自组装行为的影响应该是由于升高温度加快了水解/缩合反应的进程,有利于水解产物硅醇的生成,增强了硅醇分子间的氢键作用造成的。
4 结 论
基于超分子自组装体系的静态特征与溶胶-凝胶过程的动态性之间的对立与统一,使得溶胶-凝胶过程中有机硅氧烷的自组装行为更加复杂,从而导致前体水解/缩合行为更加复杂。尽管这方面已有相关报道,但对溶胶-凝胶过程与自组装行为之间的协同作用还有待进一步系统研究,还有着更广泛的研究空间。通过向系统中引入具有与前体强相互作用的外源分子,基于二者之间的分子间相互作用力构筑超分子自组装体系,并调整反应条件,在溶胶-凝胶过程中最大限度地保持自组装结构,将对前体水解/缩合行为及产物的长程有序结构产生重要的影响。
借助超分子自组装研究给予的丰富研究成果,结合有机硅氧烷前体水解/缩合行为特点,对于溶胶-凝胶过程中有机硅氧烷自组装行为的研究,不仅对丰富超分子自组装研究具有特别重要的理论意义,对于获得具有特殊结构和性能的硅基复合材料也具有重要的现实指导意义。
[1] Sanchez C. State of the art developments in functional hybrid materials[J].J. Mater. Chem.,2005,15:3557-3558.
[2] Schottner G. Hybrid sol-gel-derived polymers:Application of multifunctional materials[J].Chem. Mater.,2001,13(10):3422-3435.
[3] 吴超波,李伟,高大海,等.聚亚芳基有机硅氧烷的研究进展[J]. 化工进展,2008,27(8):1175-1179.
[4] Fernandes M,Cattoën X,De Zea Bermudez V,et al. Solvent-controlled morphology of lamellar silsesquioxanes:From platelets to microsponges[J].Cryst. Eng. Comm.,2011,13(5):1410-1415.
[5] Ren J,Wang L,Han X,et al. Organic silicone sol-gel polymer as a noncovalent carrier of receptor proteins for label-free Optical biosensor application[J].ACS Appl. Mater. Interfaces,2013,5(2):386-394.
[6] Loy D A,Baugher B M,Baugher C R,et al. Substituent effects on the sol-gel chemistry of organotrialkoxysilanes[J].Chem. Mater.,2000,12(12):3624-3632.
[7] Hu D D,Croutxé-Barghorn C,Feuillade M,et al. Fluorescence study of the sol-gel process in hybrid precursors:Evidence of concentration fluctuations at the local scale[J].J. Phys. Chem. B,2005,109(32):15214-15220.
[8] Kapoor P M,Yang Q,Inagaki S. Self-assembly of biphenylene-bridged hybrid mesoporous solid with molecular-scale periodicity in the pore walls[J].J. Am. Chem. Soc.,2002,124(51):15176-15177.
[9] 李祥洪. 三硅氧烷表面活性剂[J]. 有机硅材料,2004,18(1):32-34.
[10] Merkel K,Kocot A,Vij J K,et al. The orientational order parameters of a dendritic liquid crystal organo-siloxane tetrapode oligomer,determined using polarized infrared spectroscopy[J].J. Chem. Phys.,2004,121(10):5012-5022.
[11] Baney R H,Itoh M,Sakakibara A,et al. Silsesquioxanes[J].Chem. Rev.,1995,95(5):1409-1430.
[12] Novak B M. Hybrid nanocomposite materials-between inorganic glasses and organic polymers[J].Adv. Mater.,1993,5(6):422-425.
[13] Parikh A N,Allara D L,Azouz I B,et al. An intrinsic relationship between molecular structure in self-assembledN-alkylsiloxane monolayers and deposition temperature[J].J. Phys. Chem.,1994,98(31):7577-7590.
[14] Vidon S,Leblanc R M. Langmuir study of octadecyltrimethoxysilane behavior at the air-water interface[J].J. Phys. Chem. B,1998,102(7):1279-1286.
[15] Parikh A N,Schivley M A,Koo E,et al.n-Alkylsiloxanes:From single monolayers to layered crystals. The formation of crystalline polymers from the hydrolysis ofn-octadecyltrichlorosilane[J].J. Am. Chem. Soc.,1997,119(13):3135-3143.
[16] Shimojima A,Sugahara Y,Kuroda K. Inorganic-organic layered materials derivedviathe hydrolysis and polycondensation of trialkoxy (alkyl) silanes[J].Bull. Chem. Soc. Jpn.,1997,70:2847-2853.
[17] Shimojima A,Umeda N,Kuroda K. Synthesis of layered inorganic-organic nanocomposite films from mono-,di-,andtrimethoxy (alkyl) silane-tetramethoxysilane systems[J].Chem. Mater.,2001,13(10):3610-3616.
[18] Ni L,Chemtob A,Croutxé-Barghorn C,et al. Kinetics,thermodynamics,and dynamics in organosilane self-assembly[J].J. Phys. Chem. C,2012,116(45):24320-24330.
[19] Ni L,Rigolet S,Chemtob A,et al. Head-to-head and head-to-tail multilayern-alkylsilsesquioxane films[J].C. R. Chimie,2013,16(10):897-905.
[20] Fan H,Chen Z,Brinker C J,et al. Synthesis of organo-silane functionalized nanocrystal micelles and their self-assembly[J].J. Am. Chem. Soc.,2005,127(40):13746-13747.
[21] Shen S K,Sun P P,Hu D D,et al. Substituent-dominated structure evolution during sol-gel synthesis:A comparative study of sol-gel processing of 3-glycidoxypropyltrimethoxysilane and methacryloxypropyltrimethoxysilane[J].Langmuir,2010,26(11):7708-7716.
[22] Loy D A,Shea K J. Bridged polysilses-quioxanes:Highly porous hybrid organic-inorganic materials[J].Chem. Rev.,1995,95(5):1431-1442.
[23] Ariga K,Vinu A,Hill J P,et al. Coordination chemistry and supramolecular chemistry in mesoporous nanospace[J].Coordin. Chem. Rev.,2007,251:2562-2591.
[24] Cerveau G,Corriu R J P,Framery E,et al. Auto-organization of nanostructured organic-inorganic hybrid xerogels prepared by sol-gel processing:The case of a “twisted” allenic precursor[J].Chem. Mater.,2004,16(20):3794-3799.
[25] Creff G,Pichon B P,Blanc C,et al. Self-assembly of bridged silsesquioxanes:Modulating structural evolutionviacooperative covalent and noncovalent interactions[J].Langmuir,2013,29(18): 5581-5588.
[26] Fu W,He C,Jiang S,et al. Synthesis of a polymeric electron acceptor based on perylenediimide-bridged ladder polysiloxane[J].Macromolecules,2011,44(2):203-207.
[27] Dautel O J,Wantz G,Almairac R,et al. Nanostructuration of phenylenevinylenediimide-bridged silsesquioxane:From electroluminescent molecular j-aggregates to photoresponsive polymeric H-aggregates[J].J. Am. Chem. Soc.,2006,128(14):4892-4901.
[28] Moreau J J E,Vellutini L,Man M W C,et al. New hybrid organic-inorganic solids with helical morphologyviaH-bond mediated sol-gel hydrolysis of silyl derivatives of chiral (R,R)- or (S,S)-diureidocyclohexane[J].J. Am. Chem. Soc.,2001,123(7):1509-1510.
[29] Barboiu M,Cerneaux S,Van der Lee A,et al. Ion-driven ATP pump by self-organized hybrid membrane materials[J].J. Am. Chem. Soc.,2004,126(11):3545-3550.
[30] Velamakanni A,Torres J R,Ganesh K J,et al. Major controlled assembly of silane-based polymers:Chemically robust thin-films[J].Langmuir,2010,26(19):15295-15301.
[31] Tanaka M,Sawaguchi T,Kuwahara M,et al. Surface modification of silicon oxide with trialkoxysilanes toward close-packed monolayer formation[J].Langmuir,2013,29(21):6361-6368.
[32] 陈连喜,李洁,李曦,等. 高度单分散聚乙烯基倍半硅氧烷球形纳米粒子的制备及性能[J]. 高等学校化学学报,2013,34(6):1460-1465.
[33] Landskron K,Hatton B D. Periodic mesoporous organosilicas containing interconnected [Si(CH2)]3rings[J].Science,2003,302(5643):266-269.
[34] Inagaki S,Guan S,Ohsuna T,et al. An orderedmesoporous organosilica hybrid material with a crystal-like wall structure[J].Nature,2002,416(6878):304-307.
[35] Pan Y C,Wu H Y,Jheng G L,et al. Ordered and hydrothermally stable cubic periodic mesoporous organosilicas with SBA-1mesostructures:Synthesis,characterization,solid-state NMR spectroscopy,and DFT calculations[J].J. Phys. Chem. C.,2009,113(7):2690-2698.
[36] Marx S,Liron Z. Molecular imprinting in thin films of organic-inorganic hybrid sol-gel and acrylic polymers[J].Chem. Mater.,2001,13(10):3624-3630.
[37] Tanev P T,Liang Y,Pinnavaia T J. Assembly of mesoporous lamellar silicas with hierarchical particle architectures[J].J. Am. Chem. Soc.,1997,119(37):8616-8624.
[38] Park H K,Ha T H,Kim K. Gelation of octadecyltrimethoxysilane at the air/water interface:Effect of perfluorotetradecanoic acid and divalent cadmium ions[J].Langmuir,2004,20(12):4851-4858. [39] Wang S,Vidon S,Leblanc R M. Chemical and photochemical dual polymerization in a mixed langmuir monolayer of diacetylene derivatives and octadecyltrimethoxysilane[J].J. Colloid Interface Sci.,1998,207(2):303-308.
[40] Meng X J,Yokoi T,Lu D,et al. Synthesis and characterization of chiral periodic mesoporous organosilicas[J].Angew. Chem. Int. Ed.,2007,46(41):7796-7798.
[41] Silva S S,Ferreira R A S,Fu L,et al. Functional nanostructured chitosan-siloxane hybrids[J].J. Mater. Chem.,2005,15(35):3952-3961.
[42] Zhang Q,Ariga K,Okabe A,et al. A condensable amphiphile with a cleavable tail as a “lizard” template for the sol-gel synthesis of functionalized mesoporous silica[J].J. Am. Chem. Soc.,2004,126(4):988-989.
[43] Shen S K,Hu D D,Sun P P,et al. Amino acid catalyzed bulk-phase gelation of organoalkoxysilanesviaa transient cooperative self-assembly[J].J. Phys. Chem. B.,2009,113(41):13491-13498.
[44] Zhai S R,Park S S,Park M,et al. Role of inorganic salts in the formation of ordered periodic mesoporous organosilicas (PMOS) without extra acids[J].Micro. Meso. Mater.,2008,113:47-55.
[45] Zhai S,Kim I,Ha C. Synthesis and characterization of periodic mesoporous organosilicas from bridged organosilanes in the presence of mixed salts[J].J. Solid State Chem.,2008,181(27):67-74.
[46] Burleigh M C,Markowitz M A,Spector M S,et al. Porous organosilicas:An acid-catalyzed approach[J].Langmuir,2001,17(25):7923-7928.
[47] Nazli K O,Pester C W,Konradi A,et al. Cross-linking density and temperature effects on the self-assembly of SiO2-pnipaam core-shell particles at interfaces[J].Chem. Eur. J., 2013,19(18):5586-5594.
[48] Pearson T R,Warren N J,Lewis A L,et al. Effect of Ph and temperature on PMPC-PDPA copolymer self assembly[J].Macromolecules,2013,46(4):1400-1407.
[49] Pasternack R M,Amy S R,Chabal Y J. Attachment of 3-(aminopropyl)triethoxysilane on silicon oxide surfaces:Dependence on solution temperature[J].Langmuir,2008,24(22):12963-12971.
Research progress of self-assembly behavior of organosiloxane during sol-gel process
SONG Shaofei1,2,HU Daodao1,SHEN Shukun1,LI Wei1
(1School of Material Science and Engineering of Shaanxi Normal University,Xi'an 710062,Shaanxi,China;2Department of Applied Chemistry,Yuncheng University,Yuncheng 044000,Shanxi,China)
During the sol-gel process of organosiloxane,supermolecular self-assembly structures are often formed,which extremely affects the hydrolysis/condensation behavior of precursors. Mainly considering the effect of structure of alkyl group in the precursors molecules and the inducing action of additive molecules,this paper reviewes the typical studies about the self-assembly behavior of organosiloxane during the sol-gel process in recent years. The main directions in this field are analyzed. Designing and synthesizing the organosiloxane molecules with special functional group as precursors,based on the intermolecular interaction between precursors and additive molecules,to prepare the long-range ordered silica-based composite material by the cooperative action between hydrolysis/condensation reaction and supermolecular self-assembly structure will be the focus of research.
organosiloxane;hydrolysis;self-assembly;silica;composites
O 631.5
A
1000-6613(2014)08-2101-09
10.3969/j.issn.1000-6613.2014.08.027
2014-03-20;修改稿日期:2014-06-07。
国家自然科学基金委主任专项基金(21343014)及国家自然科学基金委青年科学基金(21103103)项目。
宋少飞(1979—),男,博士,讲师,主要从事有机硅氧烷溶胶-凝胶行为及功能化硅基材料制备研究。E-mail songshaofei@ ycu.edu. cn。联系人:胡道道,教授,博士生导师,主要从事有机硅氧烷簇集行为及材料可控性制备等方面研究。E-mail daodaohu@ snnu.edu.cn。
