Molecular Assessment and Taxonomic Status of the Rapid Racerunner (Eremias velox complex) with Particular Attention to the Populations in Northwestern China
2014-07-01JinlongLIUNataliaANANJEVAMarinaCHIRIKOVAKonstantinMILTOandXianguangGUO
Jinlong LIU, Natalia A. ANANJEVA, Marina A. CHIRIKOVA, Konstantin D. MILTOand Xianguang GUO
1Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, Sichuan, China
2University of Chinese Academy of Sciences, Beijing 100049, China
3Zoological Institute, Russian Academy of Sciences, St. Petersburg 199034, Russia
4Institute of Zoology, Committee of Scientif i c Ministry of Education and Science, Almaty 050060, Kazakhstan
Molecular Assessment and Taxonomic Status of the Rapid Racerunner (Eremias velox complex) with Particular Attention to the Populations in Northwestern China
Jinlong LIU1,2, Natalia A. ANANJEVA3, Marina A. CHIRIKOVA4, Konstantin D. MILTO3and Xianguang GUO1*
1Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, Sichuan, China
2University of Chinese Academy of Sciences, Beijing 100049, China
3Zoological Institute, Russian Academy of Sciences, St. Petersburg 199034, Russia
4Institute of Zoology, Committee of Scientif i c Ministry of Education and Science, Almaty 050060, Kazakhstan
The rapid racerunner, Eremias velox, is a widely distributed lizard from the northern Caucasus across entire Central Asia eastward to China. It is increasingly common to accept E. velox as a species complex in its entire range. To date, published morphological and molecular systematic hypotheses of this complex are only partially congruent, and its taxonomic status and evolutionary history are still far from clear. The mitochondrial cytochrome b gene and 12S rRNA sequences were used to evaluate the taxonomy of this complex, with particular attention to the phylogenetic placement of populations in northwestern China. Examination of the phylogenetic analyses recovers seven distinct, biogeographically discrete, and well-supported clades, revealing genetically identif i able populations corresponding to some previously morphology-def i ned subspecies. Chinese E. v. roborowskii appears to have split from other Central Asian rapid racerunner lizards well before differentiation occurred among the latter taxa. Specif i cally, we corroborate that there are two subspecies occurring in China, i.e., E. v. velox and E. v. roborowskii. We recommend a novel subspecif i c status for the phenotypically and genetically distinct populations in southern Aral Sea region of Uzbekistan previously assigned to E. v. velox. Finally, each of the three independently evolving lineages from Iranian Plateau should be recognized as three species new to science under the general lineage concept.
mtDNA, subspecies, Eremias velox complex, taxonomy, phylogeography
1. Introduction
Racerunner lizards of the genus Eremias (family Lacertidae) are the dominant reptiles in the deserts and steppes of Central Asia. The taxonomy of this genus has a long complex history, and the delimitation of species/ subspecies of Eremias is known for being notoriously diff i cult. This is, in part, caused by the great geographic variation in most morphological characters in many of the species (e.g., Szczerbak, 1974, 1975; Ananjeva et al., 1998; Chirikova, 2004). Currently, over 36 species have been assigned to the genus, and much of their phylogeny and taxonomy is controversial (e.g., Szczerbak, 1974; Guo et al., 2011). Among the many controversies, the taxonomy of the Eremias velox complex is perhaps one of the most confusing. The distribution of this group ranges from the northern Caucasus across entire Central Asia eastward to China (Figure 1). Traditionally, the rapid racerunner was considered as a single species with three or four described subspecies (Szczerbak, 1974, 1975; Eremchenko and Panf i lov, 1999). However, as noted by Rastegar-Pouyani (2009), it is increasingly common to accept E. velox as a clade in its entire range representing a species complex. The nominate form occurs in the larger parts of the distribution range. The Caspian subspecies E. v. caucasia occurs at the western part of the range. Thesubspecies E. v. roborowskii is endemic in Turpan-Hami Depression and Dunhuang Basin in northwestern China. Recently Eremchenko and Panf i lov (1999) described the fourth subspecies, E. v. borkini from the highlands of Issyk-Kul lake depression in Kyrgyzstan.
Many recent taxonomic treatments reveal varied opinions as to the number of species. Some herpetologists recognized a single widespread species (e.g., Szczerbak, 2003; Ananjeva et al., 2006; Sindaco and Jeremcenko, 2008), whereas others (Rastegar-Pouyani, 2009; Rastegar-Pouyani et al., 2012) hypothesized the existence of at least four distinct species and three well-supported subspecies (Figure 2). Some of the differences of opinion result from the accumulation of mitochondrial DNA sequence information (Rastegar-Pouyani et al., 2012). At present it seems reasonable that mtDNA sequence information supports the recognition of the Iranian populations of E. velox as three distinct species based on differences in distribution, morphology, habitat preference, and mtDNA sequence divergence (Figure 2).
The subspecific status of E. v. roborowskii is still controversial, although Szczerbak (1975) confirmed its status according to color, pattern and characters of pholidosis. Boulenger (1921) suggested that the morphological variation in the populations of E. v. roborowskii is not sufficient to support its subspecies status. The validity of this subspecies has not been recognized for a long time (Mertens and Wermuth, 1960). More recently, on the basis of multivariate morphological analyses, Wang et al. (2014) argued that there is no subspecies differentiation between populations from Turpan-Hami Depression and from Dzungarian Basin and Ily Valley in China. Thus, Wang et al. (2014) implied that E. v. roborowskii may be a synonym of nominate subspecies.
Establishing correct species/subspecies limits depends on adequate sampling of populations throughout the range. Given the previous studies, it might appear that taxonomic limits were clear. Rastegar-Pouyani (2009) made the first attempt to elucidate the phylogenetic relationships among the rapid racerunner in Iran and Central Asia on the basis of ISSR-PCR fingerprints data. The results were tentative due in part to limited taxonomic sampling. As shown by simulations and empirical studies, more taxa and data do affect the phylogenetic reconstruction (e.g., Pollock et al., 2002; Zwickl and Hillis, 2002). Rastegar-Pouyani et al. (2012) studied patterns of mitochondrial DNA cytochrome b (cyt b) and 12S rRNA variation in 70 specimens from 13 geographically distant localities in Iran and Central Asia. Their results contradicted previous systematic hypotheses; revealed surprising relationships between Iranian and Central Asiaian lineages and uncovered novel, cryptic evolutionary lineages (i.e. new putative species). Moreover, they argued that the eastern and southern Kazakhstan populations correspond to the traditional E. v. roborowskii. However, Rastegar-Pouyani et al. (2012), who used most samples already analyzed by Rastegar-Pouyani (2009), were unable to sample adequately the eastern part of the taxon’s range. There was no sample from China included in their work. In addition, Rastegar-Pouyani et al. (2012) speculated that E. v roborowskii occurs in western China, eastern Kazakhstan and Eastern Uzbekistan. This inappropriate interpretation may give confusing clues about the identity of this subspecies, resulting in incorrect inference of its phylogenetic placement. It appears likely that the inclusion of more rapid racerunner samples may get a better illustration of their phylogeographic structure, and may also provide valuable information for their taxonomy.

Figure 1 Entire distribution range of Eremias velox complex showing traditional subspecies’ range with different background color, and the sampling localities for different populations (numerical labels 14–30 with green dots), white dots with numbers 1–13 representing those from Rastegar-Pouyani et al.(2012). The locality number corresponds with that in Appendix A.
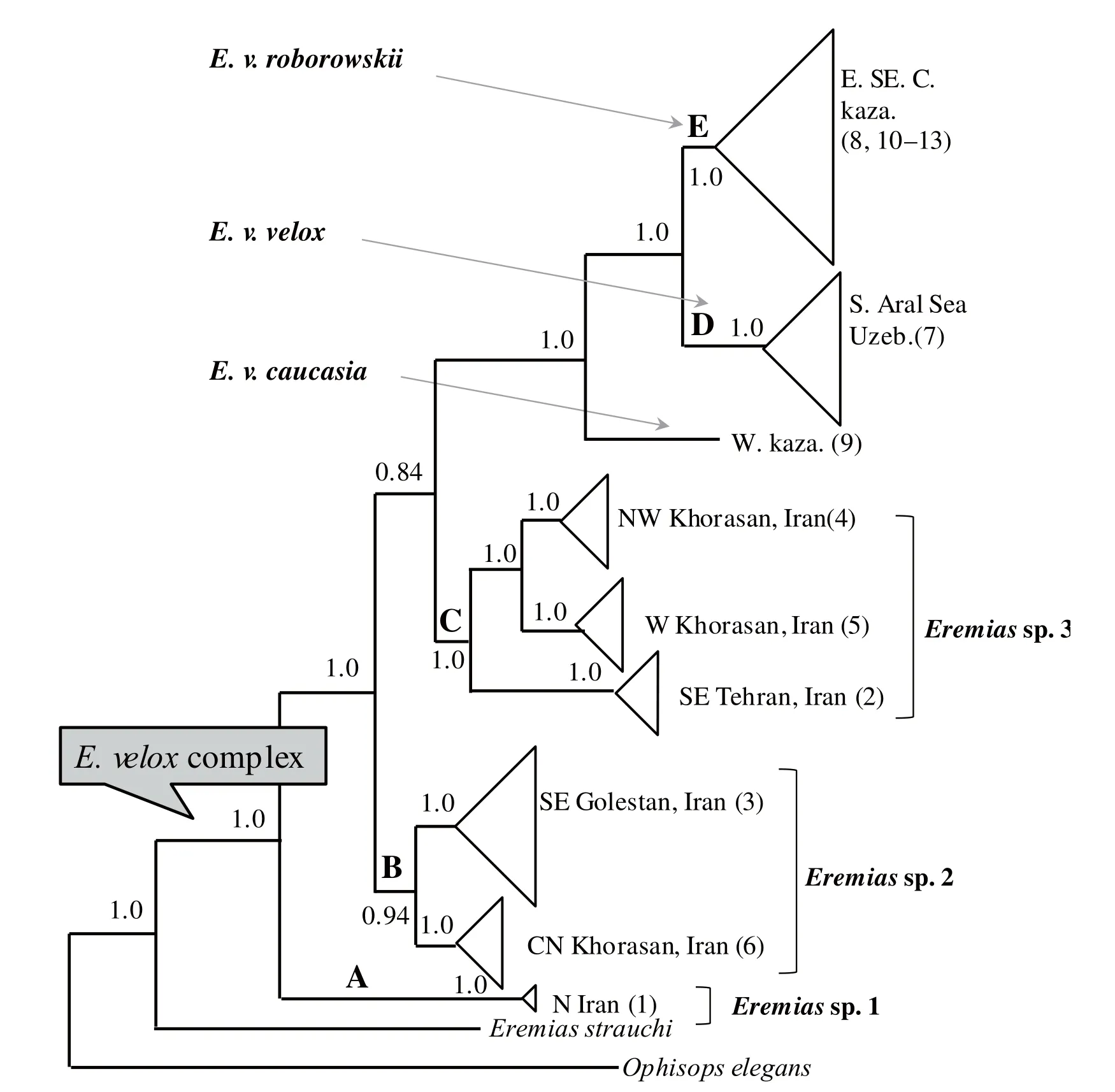
Figure 2 Rastegar-Pouyani et al.’s (2012) phylogenetic hypothesis for populations of the Eremias velox complex from Iranian Plateau and Central Asia using partitioned Bayesian analyses of mtDNA data. A, B, C, D and E indicate the major clades. Values beside the node represent Bayesian posterior probabilities.
Thus, as an extension of the molecular phylogenetics of the rapid racerunner, an effort has been made to collect some specimens from northwestern China and Russia. We further determined the complete cyt b gene and 12S rRNA segment to gain a more comprehensive view on phylogeographic patterns in E. velox complex. This study specif i cally aims to: (1) re-evaluate the phylogeographic structure of E. velox complex, (2) test the phylogenetic affinities of the rapid racerunner populations in China, and (3) test the validity of the subspecif i c status of E. v. roborowskii. The hypothesis that E. v. roborowskii is valid particularly predicts that it will correspond with distinct, well-supported, and moderately divergent mtDNA haplotype clade.
2. Materials and Methods
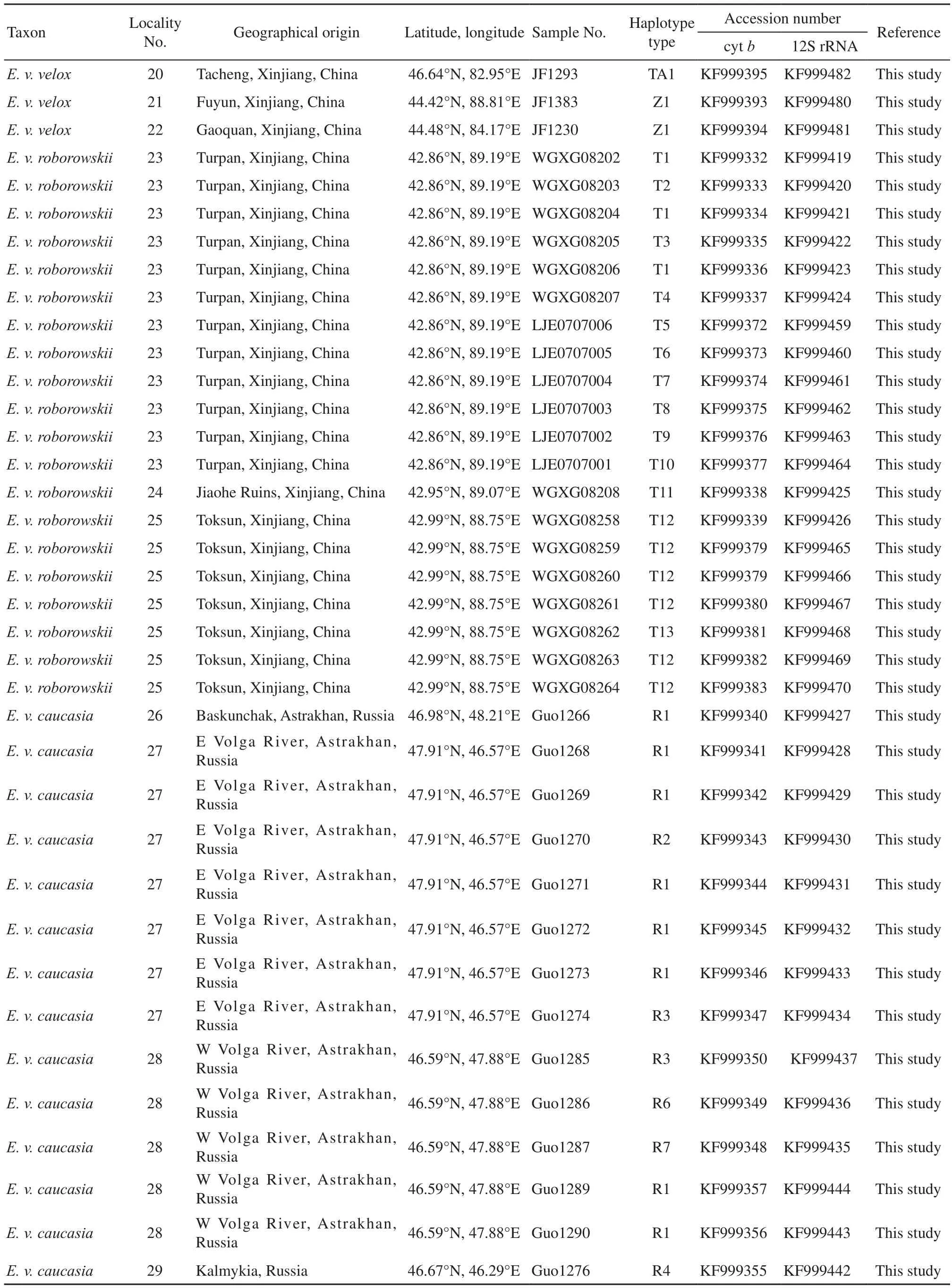
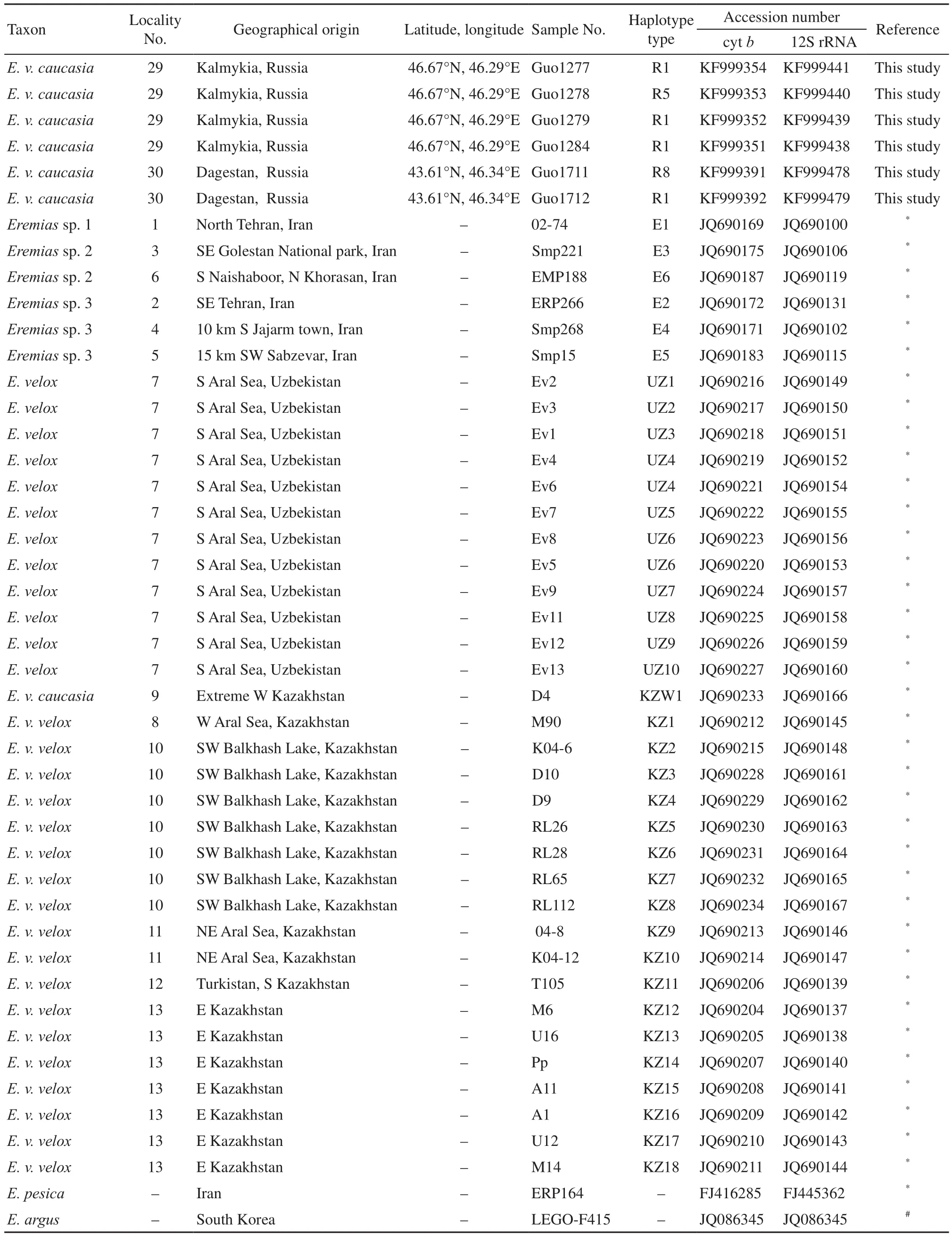
2.1 Taxon samplingWe collected a total of 87 samples representing natural populations at 17 localities. These included 57 samples from 12 localities in China, 30samples from five localities in Russia, covering a large territory occupied by E. velox in the two regions (Appendix A, Figure 1). To ensure accuracy more than four representatives of each population were collected. However, only a single individual was collected from six sites due in part to sampling diff i culties. Populations were assigned subspecific status based on location and phenotype. Due to logistical and legal obstacles to filed work, no sample of E. velox borkini from the highlands of Kyrgyzstan was available. Based on present knowledge of phylogenetic relationships among Eremias lizards, E. argus and E. persica were chosen as outgroup taxa (Guo et al., 2011). All voucher specimens were deposited in the herpetological collections of Chengdu Institute of Biology, Chinese Academy of Sciences or the Department of Zoology, University of Guelph. Detailed information about all specimens and sequences used in this study is listed in Appendix A.
2.2 DNA extraction, amplification, and sequencingGenomic DNA was extracted from liver or tail muscle tissues stored in 95% ethanol following the protocol of Aljanabi and Martinez (1997). Complete sequence of the mitochondrial DNA cyt b gene was amplified using the primers L14919 and H16064 from Burbrink et al.(2000). A ~371bp fragment of 12S rRNA was amplif i ed using the primers L1091 and H1478 from Kocher et al.(1989). Each of our 50 μl PCR reactions contained 25 μl of 2 × EasyTaq SuperMix, 0.4 μM of each primer, and 1-2 μl genomic DNA. The PCR protocol involved initial denaturation at 94°C for 180 s followed by 30 cycles of 94°C for 30 s, 43°C for 30 s (50–52°C for 12S rRNA), and elongation at 72°C for 90 s (60 s for 12S rRNA); and f i nal extension at 72°C for 8 min. Negative controls were run for all amplif i cations. Amplif i ed products were visualized on 0.8% agarose gels and purified using the DNA gel extraction kit (Omega Bio-Tek). The purified products were sequenced directly by Majorbio or Sangon (Shanghai) Co., Ltd., using the same primers as PCR for 12S rRNA, and L14919 from Burbrink et al. (2000) in combination with L14761 as well as H14892 from Zhao et al. (2011) for cyt b. All novel sequences are deposited in GenBank with accession numbers KF999318–KF999491.
2.3 Sequence alignment and analysesWe have retrieved sequences of cyt b and 12S rRNA of 39 individuals from GenBank, which included sequences of 37 individuals published in Rastegar-Pouyani et al. (2012) and sequences of two individuals used as outgroup in this study (Appendix A). Sequences of cyt b were translated to amino acid sequences using MEGA5 (Tamura et al., 2011) to verify the data. A total of 126 sequences of cyt b and 12S rRNA were first aligned using Clustal X 2.0 (Larkin et al., 2007), respectively, with default gap penalties. Then the aligned matrice were re-checked visually.
Phylogenetic congruence of cyt b and 12S rRNA data sets were tested by the partition homogeneity test of Farris et al. (1995) with PAUP* 4.0b10 (Swofford, 2003). The partition homogeneity test supported the combination of the cyt b and 12S rRNA data sets (P = 0.1). Homogeneity of base frequencies was evaluated using Chi-square (χ2) tests implemented in PAUP*. Base composition at the third position exhibited no heterogeneity: the combined data, χ2= 16.66, df = 237, P = 1.00; first position, χ2= 7.18, df = 237, P = 1.00; second position, χ2= 2.02, df = 237, P = 1.00; third position, χ2= 48.99, df = 237, P = 1.00; and 12S rRNA, χ2= 12.11, df = 237, P = 1.00. Given these results, a sensible approach to analyze these data phylogenetically would be to apply a time-reversible Markov model.
2.4 Phylogenetic analysesTo estimate phylogenetic relationships, Bayesian inference (BI), maximum likelihood (ML), and maximum parsimony (MP) were implemented for the combined data. Each haplotype was treated as a taxon in the analyses. MP analyses with PAUPRat (Sikes and Lewis, 2001), based on Parsimony Ratchet (Nixon, 1999), were conducted using 1000 ratchets with 10 iterations per replicate. The shortest equally most parsimonious trees were combined to produce a strict consensus tree. Consistency (CI), retention (RI), and rescaled consistency (RC) indices were used to evaluate homoplasy in character data and overall support for optimal trees. Support for individual branches was evaluated using non-parametric bootstrapping (Felsenstein, 1985) in PAUP* with 1000 pseudoreplicates, TBR branch swapping, simple taxon addition with one tree held at each step, and a maximum of 100 trees saved per replicate in order to decrease the time needed to run large bootstrap replicates.
To infer relationships under BI we used MrBayes v3.2.2 (Ronquist et al., 2012). We optimized several parameters prior to conducting our final analyses using the Markov Chain Monte Carlo (MCMC) method implemented in MrBayes, including partitioning strategy, models of molecular evolution, length of MCMC analyses. We assessed two alternative partitioning strategies: (1) a single partition and (2) a four-partition strategy that applied a single partition to each of the three codon positions of cyt b and a fourth partition for 12SrRNA sequence.
The Akaike Information Criterion (AIC; Akaike, 1974), as implemented in MrModeltest v2.3 (Nylander, 2008), was used to select the appropriate model of sequence evolution for each partition. To compare the two alternative partitioning strategies, we conducted two independent analyses of each strategy in MrBayes. Each analysis consisted of three heated chains and one cold chain, with an incremental heating temperature of 0.02, and an exponential distribution with a rate parameter of 25 as the prior on branch lengths (Marshall, 2010). All analyses were run for 20 million generations with parameters and topologies sampled every 1000 generations. To ensure MCMC convergence, a burn-in period for all analyses was diagnosed in two ways: (1) by the average standard deviation of split frequencies (ASDSF; Lakner et al., 2008) between two MCMC analyses run independently, with levels below 0.01 being considered indicative of convergence, and (2) by the potential scale reduction factor (PSRF; Gelman and Rubin, 1992) comparing the variance within and between runs, with levels approaching 1.0 as runs converge. To compare performance of the two partitioning strategies we calculated Bayes factors from the marginal likelihood scores estimated by the stepping-stone method (Xie et al., 2011) in MrBayes 3.2.2. Comparison of alternative partitioning strategies strongly favored four partitions over one partitioning scheme (2Ln Bayes factor > 600). After selecting the optimal partitioning strategy, we conducted two analyses with different starting trees using the optimized parameters. We then used the sumt command in MrBayes to generate a consensus topology and associated Bayesian posterior probabilities (BPP) from a pooled post-burnin sampled from two analyses.
Partitioned maximum likelihood (ML) analyses were conducted in RAxMLHPC v7.0 (Stamatakis, 2006) on the combined dataset with the same partitioning strategy as for Bayesian inference. The more complex model (GTR + I + Γ) was used for all subsets, and 100 replicate ML inferences were performed for each analysis. Each inference was initiated with a random starting tree and nodal support was assessed with 1000 bootstrap pseudoreplicates (Stamatakis et al., 2008).
2.5 Bayesian tests of topology hypothesesBayes factors were used to compare the unconstrained Bayesian tree topology to Bayesian trees with ‘hard’constraints. Here we tested the following taxonomy and geography-based hypotheses to evaluate the taxonomic status within E. velox complex: (1) Are populations within the historically accepted distributions in northwestern China monophyletic? (2) Are the Iranian populations monophyletic? (3) Do Iranian populations belong to nominate subspecies? (4) Are populations of E. v. roborowskii and nominate subspecies monophyletic? We constrained the topology for each of the above putative groupings and performed a stepping stone run of 10 million generations (50 steps with convergence being reached in each step) from which we obtained a mean marginal likelihood estimate. Convergence was examined through diagnostic plots of standard deviation of the split frequencies and screening similarity in the two independent marginal likelihood estimates (Ronquist et al., 2011). Each of these estimates were compared using Bayes factors (Kass and Raftery, 1995) to the marginal likelihood estimate obtained from the unconstrained MrBayes run of the same length. The stepping stone function (Xie et al., 2011) implemented in MrBayes v3.2.2, offers an improved estimation of marginal likelihood over harmonic mean estimation.
3. Results
3.1 Sequence characteristicsTwo mitochondrial genes (cyt b and 12S rRNA) were obtained, yielding alignments of 1143 and 372 bp, respectively. 126 sequences (including two outgroup taxa) revealed 80 haplotypes. Alignment was unambiguous and 1 indel was inferred in 12S rRNA between outgroup and ingroup taxa. No premature stop codons were observed in cyt b, indicating that the obtained sequences were mitochondrial in origin and not nuclear pseudocopies. Average base composition is A: 27.55%, C: 30.69, G: 14.72%, T: 27.04%, when the outgroup taxa are combined. Nucleotide composition showed an anti-G bias and A+T-richness (54.59%), which is a characteristic of the mitochondrial genome. Variable and parsimony-informative characters are: 441 and 319 of 1143 (cyt b); 84 and 57 of 371 (12S rRNA).
A χ2test at the 5% level of signif i cance for differences in base frequencies showed that there was no base compositional heterogeneity among sequences, which is known to adversely affect phylogenetic inference (Jermiin et al., 2004).
3.2 Phylogenetic analysesThe heuristic search of the combined data resulted in 914 equally parsimonious trees of 1136 steps, with moderate CI (0.579) and high RI (0.887). These are presented as a majority-rule consensus tree where branches with bootstrap support lower than 50% are collapsed (data not shown). AIC analyses conducted with the aid of MrModeltest identified theHKY + I + Γ model as the most appropriate model for partition of cyt b 1stcodon position, and the GTR + I + Γ model for partitions of cyt b 2ndcodon position, cyt b 3rdcodon position, 12S rRNA or combined data. Bayesian analyses were conducted for 20 million generations, with a conservative burn-in of six million generations that provided dense sampling of the posterior distribution. The MP, ML and Bayesian trees differed in support values, but not in the compositions of the major lineages. The bootstrap support was also marked on the branches that receive such support in the Bayesian tree (Figure 3). The phylogenetic trees recovered the monophyly of the E. velox complex with high support values. As shown in Figure 3, seven major clades connected deep in the complex phylogenetic history were identified in all reconstructions with strong posterior probabilities and bootstrap values, which generally correspond well to geographical regions within the distribution range. These clades were designated I, II, III, IV, V, VI and VII in Figure 3. The subdivision and phylogenetic positions of the Iran populations (I, II, III) are completely identical with the clades A, B, C of Rastegar-Pouyani et al. (2012), respectively. The basal split in the complex is between the northern Iranian population (clade I) and a highly supported clade of the other populations from Iran, Central Asia, China and Russia. However, clade IV, corresponding to E. v. roborowskii, covers the populations from Turpan-Hami Depression, and forms the sister taxon to the other populations excluding those from Iran. The clade V, corresponding to E. v. caucasia, covers populations from North-eastern Russian Caucasus and Western Kazakhstan. The clade VI is composed of populations from Uzbekistan, conventionally assigned to nominate subspecies. The clade VII covers populations from Eastern Kazakhstan and Dzungarian Basin as well as Ily Valley in China, which corresponds to nominate subspecies. Overall, the notable differences between Rastegar-Pouyani et al. (2012) and our results presented here lie in the phylogenetic placement of E. v. roborowskii.
3.3 Bayesian tests of topology hypothesesWe compared the marginal likelihood estimates of topologically constrained and unconstrained run of MrBayes for possible monophyletic group within E. velox complex (Table 1). In all cases, there was very strong (Bayes factor > 100) evidence against the constrained topologies. Thus, four alternative phylogenetic hypotheses were significantly rejected, such as a monophyletic group of Chinese rapid racerunner populations, monophyly of Iranian populations, assignment of Iranian populations to nominate subspecies, and assignment of E. v. roborowskii to nominate subspecies.
4. Discussion
This study provided strong support for the monophyly of E. velox, consistent with the results of Guo et al. (2011) and Rastegar-Pouyani et al. (2012), and thus providing a fi rm basis for future testing of alternative biogeographic models. The Iranian populations were recovered here as a paraphyletic assemblage, representing three distinct, biogeographically discrete, well-supported clades that are not each other’s closet relatives. This is in accord with the results of Rastegar-Pouyani et al. (2012).
The morphological differences (Rastegar-Pouyani et al., 2012), allopatric ranges and substantial genetic divergence in the mtDNA bear evidence of long standing isolation between several populations. The haplotype clades associated with each lineage on Iranian Plateau are differentiated by mean uncorrected levels of sequence divergence that often exceed 10% (Table 2). This degree of divergence suggests that evolutionary separation of the three clades occurred millions of years ago, likely sometime during the Miocene based on a molecular clock calibration of 1.25% pairwise divergence per million years for mtDNA within the E. velox complex (Rastegar-Pouyani et al., 2012). Concordance between strongly differentiated mtDNA haplotype clades and phenotypicvariation (Rastegar-Pouyani et al., 2012) supports the hypothesis that each of the three independently evolving lineages from Iranian Plateau deserves recognition at the species-level under the general lineage concept of species (de Queiroz, 1999; 2007) and associated operational species-delimitation criteria (Wiens and Penkrot, 2002).

Table 1 Bayes factors of stepping-stone-based estimates of marginal likelihood for comparisons of alternative phylogenetic hypotheses. 2ln Bayes factors ≥10 are considered very strongly different (Kass and Raftery, 1995), indicating evidence against alternative hypotheses.
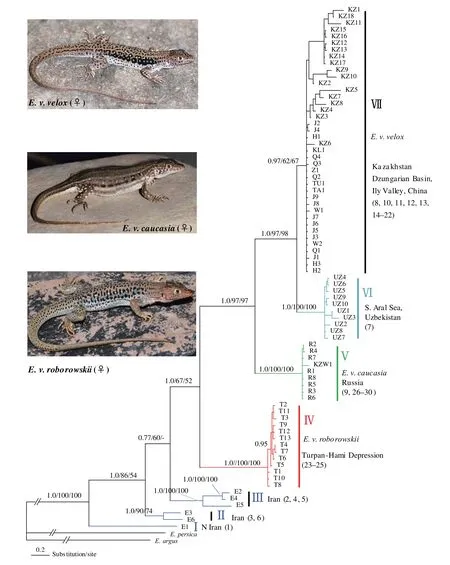
Figure 3 Hypothesized relationships of Eremias velox complex included in this study, illustrated by the majority-rule consensus tree resulting from partitioned Bayesian analyses. Bayesian posterior probabilities, maximum parsimony and likelihood bootstrap values are shown. Nodes supported by ≥ 0.95 Bayesian PP and ≥ 70% BP were considered highly supported. Terminals are labeled with haplotypes. Roman alphabet labels correspond to clades referred to the Results and Discussion. Representative photographs of female adults show the dorsal and lateral differentiation among three subspecies.
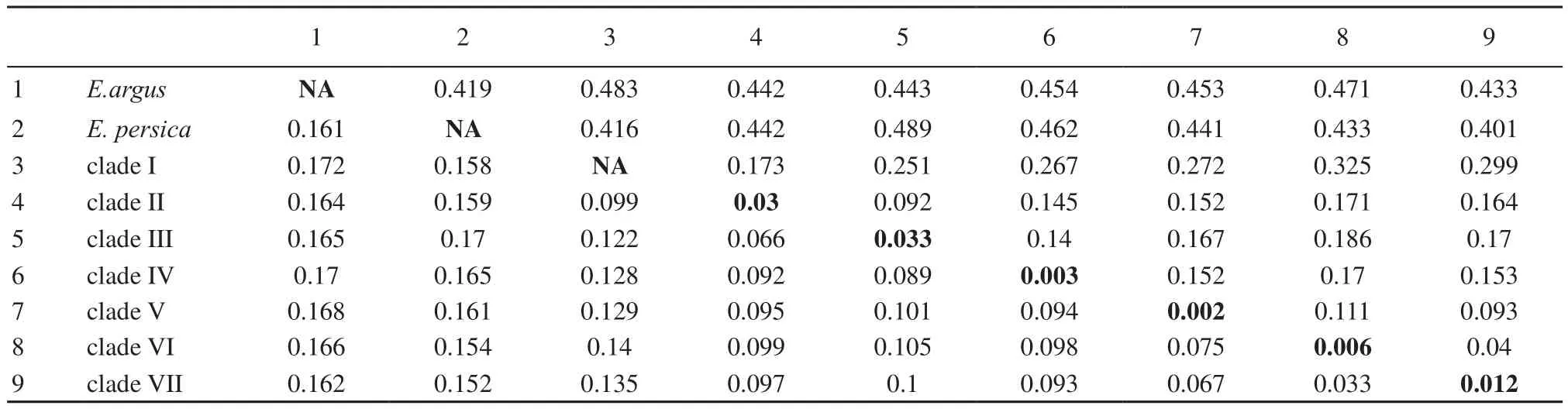
Table 2 Genetic distances within and between outgroup species and E. velox complex included in this study. Mean uncorrected (p) distances are below diagonal; distances calculated using MEGA's Maximum Composite Likelihood model with rate variation among sites def i ned by the gamma shape parameter (0.2366) are above the diagonal. Mean maximum likelihood divergences within each species or subspecies are in the diagonal with bold text.
Rastegar-Pouyani et al. (2012) demonstrated the basal positions of the Iranian clades for the first time and considered them three distinct species. However, their inference of the phylogenetic placement of E. v. roborowskii was misleading. In support of Rastegar-Pouyani et al. (2012), our results confirm the basal position of the Iranian clades and statistically reject the placement of Iranian clades into the nominate subspecies from Central Asia. Surprisingly, we find that E. v. roborowskii is the sister taxon to E. velox caucasia plus E. v. velox, i.e., other populations from Central Asia. Our results clearly confirm that all of the major clades inferred from mitochondrial haplotypes are concordant with geographical regions. Iranian clades at the basal position of the phylogenetic tree may imply that E. velox originated from the Iranian Plateau and invaded Central Asia as Rastegar-Pouyani et al. (2012) suggested. If we take this scenario as a reliable explanation for the origin and evolution of E. velox, we consider the clade IV (E. v. roborowskii) as first split from entire Central Asian populations, resulting from the barrier effect of Tianshan Mountains with their dramatic uplift since Tertiary (Macey et al., 1999; Guo and Wang, 2007; Zhang et al., 2008; Melville et al., 2009; Solovyeva, 2013). Geographically, with the uplift of Tianshan Mountains, Turpan-Hami Depression and Dzungarian Basin were blocked from each other, resulting in a historical vicariant pattern and allopatric distribution between E. v. roborowskii and E. v. velox as well as E. v. caucasia. Wang et al. (2014) failed to recognize the validity of E. v. roborowskii due to a lack of understanding the discrete characters between E. v. velox and E. v. roborowskii, including dorsal pattern and lateral color pattern. In general, the adults of E. v. roborowskii have a dorsal pattern of irregular dark spots, with rows of bright blue-edged, dark eye-like spots on the lateral sides (Zhao, 1999; Szczerbak, 2003).
In addition, clade V, corresponding to traditional E. velox caucasia, forms the sister taxon to Central Asian clades, in accord with the results of Dolotovskaya et al.(2007) inferred from ~ 1800 bp mtDNA of cyt b, COI and 16S rRNA . Rastegar-Pouyani et al. (2012) speculated that a single specimen from Western Kazakhstan belongs to E. velox caucasia. The affinity of that single specimen was corroborated in this study for there to be strong support for its nesting inside clade V. The Central Asian clades, corresponding to conventional nominate subspecies, comprise clades VI and VII. The patterns of mtDNA variation support a sister relationship between clade VI and clade VII. Clade VI comprises samples from Uzbekistan, whereas clade VII consists of populations from Kazakhstan and Dzungarian Basin and Ily River Valley, Xinjiang, China. Judging from the type locality of the nominate form of E. velox, Inderskija mountain in Kazakhstan, we consider clade VII as nominate subspecies. Szczerbak (1975) examined the relationships of E. v. roborowskii from Ssatschsheu (Dunhuang Basin, Gansu, China) and Lukuchun (Shanshan, Xinjiang, China) with a population from Dzungarian Basin. He found that the samples from Dzungarian Basin stood an intermediate position between the subspecies E. v. roborowskii and nominative form. On the one hand, judging from pholidosis, the population from Dzungarian Basin is closer to E. v. roborowskii than to the populations from Balkhash in Kazakhstan. On the other hand, the dorsal and lateral pattern of the population from Dzungarian Basin is closer to that of the latter, i.e. nominate form.Interestingly, Szczerbak (1975) referred to the population from Dzungarian Basin as nominative form. Our results statistically support that the rapid racerunner populations in Dzungarian Basin cluster with those from Kazakhstan. In fact, biogeographic connections to the Dzungarian Basin are to the northwest, with a number of passes leading to Kazakhstan. The Dzungarian Gate is the main pass through the western ranges, leading to Lake Alaköl and Lake Balkhash in Kazakhstan. There are a number of taxa that have similar distribution pattern, including Phrynocephalus melanurus (Melville et al., 2009), P. heliocopus (Solovyeva et al., 2011), and Eremias arguta (Szczerbak, 2003).
Traditionally, the subspecies category is used to diagnose geographically distinct populations that were thought to be in the early stages of speciation (Mayr, 1963; Mayr and Ashlock, 1991). Despite some criticisms surrounding the concept of subspecies (e.g., Burbrink et al., 2000; Patten and Unitt, 2002; Zink, 2004), recent studies in which researchers used multiple criteria (e.g., morphological, behavioral, and genetic characters) have confirmed that many subspecies are evolutionarily definable entities (e.g., Gavin et al., 1999; Phillimore and Owens, 2006; Guo et al., 2012). In Central Asia, the clinal and discrete variation among E. velox populations from different regions have been documented (Szczerbak, 1975; Chirikova, 2004). As noted by Szczerbak (1975), the most deviating populations of E. velox were found in Fergana Valley and neighborhood of Badkhyz desert, reaching subspecies level. Chirikova (2004) studied geographic variations of E. velox populations from Kazakhstan and Uzbekistan. She found some variations in phenotypical traits between populations from the two regions. In contrast with the nominate form in Kazakhstan, populations from the southern Aral Sea region in Uzbekistan have very light elongated spots on the middle of the back in adults. Even some individuals from the southern Aral Sea region lack this kind of dorsal pattern completely. Our results demonstrate that the populations in southern Aral Sea region of Uzbekistan correspond with distinct, well-supported, and moderately divergent mtDNA haplotype clade (clade VI) . The mean uncorrected level of sequence divergence between clades VI and VII falls within 3.3%. In support of Chirikova (2004), we recommend that the populations of rapid racerunner from southern Aral Sea region of Uzbekistan be assigned to a novel subspecies, which will facilitate an effective short-cut for estimating patterns of intraspeci fi c genetic diversity of E. velox.
5. Conclusions
The mtDNA data confirms the subspecific status for three of the four morphology-defined subspecies: E. v. roborowskii, E. v. caucasia, E. v. velox (sensu stricto). Specifically, we corroborate that there are two subspecies occurring in China, i.e., E. v. velox and E. v. roborowskii. We recommend a novel subspecies for the phenotypically and genetically distinct populations in Uzbekistan previously referred to as E. v. velox. We also support species-level recognition for each of the three independently evolving lineages from Iranian Plateau traditionally assigned to E. v. velox. We do so recognizing that more comprehensive geographic sampling and investigation of additional molecular markers are required to clarify hypothesized species/subspecies boundaries, taxonomic status and evolutionary history of the E. velox complex.
Acknowledgements We wish to offer our sincere thanks to everyone who helped make this paper a reality. We are grateful to Jinzhong FU and Yin QI for providing some tissue samples in their care. We extend our gratitude to Yuezhao WANG, Qiang DAI, Jun LI, Hongfu WAN, Bo CAI, Daniel A. MELNIKOV, Nikolai N. ORLOV, and Konstantin LOTIEV for assistance in collecting specimens. We thank Tatjana N. DUJSEBAYEVA for her helpful comments and Kerry A. COBB (The University of Kansas) for his critical reading of an earlier version of this manuscript. This research was supported by the National Natural Science Foundation of China (31272281), Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-Q-6), and Projects of the Bureau of International Co-operation of the Chinese Academy of Sciences (No. 1136, 1327, 1441).
Akaike H. 1974. A new look at the statistical model identif i cation. IEEE Trans Automat Contr, 19: 716–723
Aljanabi S. M., Martinez I. 1997. Universal and rapid saltextraction of high quality genomic DNA for PCR-based techniques. Nucl Acids Res, 25: 4692–4693
Ananjeva N. B., Borkin L., Darevsky I. S., Orlov N. L. 1998. Amphibians and Reptilies. Encyclopedia of the Nature of Russia (Field Guide of Amphibians and Reptiles of Russia and Adjacent Countries). Moscow: ABF Publishing Company, 574 pp (In Russian)
Ananjeva N. B., Orlov N. L., Khalikov R. G., Darevsky I. S., Ryabov I. S., Barabanov A. V. 2006. The Reptiles of North Eurasia: Taxonomic Diversity, Distribution, Conservation Status. Bulgaria: Pensoft Publishers, 250 pp
Boulenger G. A. 1921. Monograph of the Lacertidae, Vol. II. London: Trustees of the British Museum of Natural History, VIII + 451 pp
Burbrink F. T., Lawson R., Slowinski J. B. 2000. Mitochondrial DNA phylogeography of the polytypic North American rat snake (Elaphe obsoleta): A critique of the subspecies concept. Evolution, 54: 2107–2118
Chirikova M. A. 2004. Variability of Eremias velox Pallas, 1771 (Reptilia, Sauria) from Kazakhstan. Selevinia, 24–34 (In Russian with English abstract)
de Queiroz K. 1998. The general lineage concept of species, species criteria, and the process of speciation: a conceptual unif i cation and terminological recommendations. In Howard D. J., Berlocher S. H. (Eds), Endless Forms: Species and Speciation. Oxford: Oxford University Press, 57–75
de Queiroz K. 2007. Species concepts and species delimitation. Syst Biol, 56: 879–886
Dolotovskaya S. I., Chirikova M. A., Solovyeva E. N., Poyarkov N. A., Wan L., Orlova V. F. 2007. Molecular and morphological differentiation of rapid racerunner Eremias velox (Lacertidae) with comments on taxonomy and biogeography of Middle-Asian racerunners. Abstracts of 14thEuropean Congress of Herpetology and SEH Ordinary General Meeting, Porto, Portugal, 203
Eremchenko V., Panfilov A. 1999. Taxonomic position and geographic relations of a lacertid lizard Eremias velox from the Issyk-Kul lake depression, Tien Shan Mountains, Kyrgyzstan. Science and New Technology, 99: 119–125
Felsenstein J. P. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39: 783–791
Frankham R., Ballou J. D., Briscoe D. A. 2002. Resolving taxonomic uncertainties and defining management units. In Frankham R., Ballou J. D., Briscoe D. A. (Eds), Introduction to Conservation Genetics. Cambridge, UK: Cambridge University Press, 365–392
Gavin T. A., Sherman P. W., Yensen E., May B. 1999. Population genetic structure of the northern Idaho ground squirrel (Spermophilus brunneus brunneus). J Mammal, 80: 156–168
Guo X., Dai X., Chen D., Papenfuss T. J., Ananjeva N. B., Melnikov D. A., Wang Y. 2011. Phylogeny and divergence times of some racerunner lizards (Lacertidae: Eremias) inferred from mitochondrial 16S rRNA gene segments. Mol Phylogenet Evol, 61: 400–412
Guo X., Liu L., Wang Y. 2012. Phylogeography of the Phrynocephalus vlangalii species complex in the upper reaches of the Yellow River inferred from mtDNA ND4-tRNALEUsegments. Asian Herpetol Res, 3: 52–68
Guo X., Wang Y. 2007. Partitioned Bayesian analyses, dispersalvicariance analysis, and the biogeography of Chinese toadheaded lizards (Agamidae: Phrynocephalus): A re-evaluation. Mol Phylogenet Evol, 45: 643–662
Jermiin L., Ho S. Y., Ababneh F., Robinson J., Larkum A. W. 2004. The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst Biol, 53: 638–643
Kass R. E., Raftery A. E. 1995. Bayes factors. J Am Statist Assoc, 90: 773–795
Kocher T. D., Thomas W. K., Meyer A., Edwards S. V., Pääbo S., Villablanca F. X., Wilson A. C. 1989. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci, 86: 6196–6200
Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thomson J. D., Gibson T. J., Higgins D. G. 2007. Clustal W and Clustal X version 2.0. Bioinformatics, 23: 2947–2948
Macey J. R., Wang Y., Ananjeva N. B., Larson A., Papenfuss T. J. 1999. Vicariant patterns of fragmentation among gekkonid lizards of the genus Teratoscincus produced by Indian collision: a molecular phylogenetic perspective and an area cladogram for Central Asia. Mol Phylogenet Evol, 12: 320–332
Marshall D. C. 2010. Cryptic failure of partitioned Bayesian phylogenetic analyses: Lost in the land of long trees. Syst Biol, 59: 108–117
Mayr E. 1963. Animal Species and Evolution. Cambridge, Massachusetts: Belknap Press, XVI + 797 pp
Mayr E., Ashlock P. D. 1991. Principles of Systematic Zoology, second edition. New York: McGraw-Hill, XX + 475 pp
Melville J., Hale J., Mantziou G., Ananjeva N. B., Milto K., Clemann N. 2009. Historical biogeography, phylogenetic relationships and intraspecific diversity of agamids lizards in the Central Asian deserts of Kazakhstan and Uzbekistan. Mol Phylogenet Evol, 53: 99–122
Mertens R., Wermuth H. 1960. Die Amphibien und Reptilien Europas. Verlag Waldemar Kramer, Frankfurt am Main. XII + 264 pp
Nixon K. C. 1999. The Parsimony Ratchet, a new method for rapid parsimony analysis. Cladistics, 15: 407–414
Nylander J. A. A. 2008. MrModeltest 2.3. Program distributed by the author. Evolutionary Biology Centre, Uppsala University
Patten M. A., Unitt P. 2002. Diagnosability versus mean differences of sage sparrow subspecies. Auk, 119: 26–35
Phillimore A. B., Owens I. P. F. 2006. Are subspecies useful in evolutionary and conservation biology? Proc R Soc B, 273: 1049–1053
Pollock D. D., Zwickl D. J., Mccguire J. A., Hillis D. M. 2002. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol, 51: 664–671
Rastegar-Pouyani E. 2009. The phylogeny of the Eremias velox complex of the Iranian Plateau and Central Asia (Reptilia, Lacertidae): Molecular evidence from ISSR-PCR fingerprints. Iranian J Animal Biosystemat, 5: 33–46
Rastegar-Pouyani E., Kazemi Noureini S., Rastegar-Pouyani N., Joger U., Wink M. 2010. Molecular phylogeny and biogeography of the Eremias persica complex of the Iranian Plateau (Reptilia: Lacertidae) based on sequences of the mtDNA. Zool J Linn Soc, 158: 641–660
Rastegar-Pouyani E., Kazemi Noureini S., Rastegar-Pouyani N., Joge, U., Win, M. 2012. Molecular phylogeny and intraspecif i c differentiation of the Eremias velox complex of the Iranian Plateau and Central Asia (Sauria, Lacertidae). J Zool Systemat Evol Res, 50: 220–229
Ronquist F., Huelsenbeck J. P., Teslenko M. 2011. Draft MrBayes version 3.2 manual: Tutorial and model summaries
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., Larget B., Liu L., Suchard M. A., HuelsenbeckJ. P. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol, 61: 539–42
Sikes D. S., Lewis P. O. 2001. PAUPRat: PAUP* implementation of the parsimony ratchet. Beta software, version 1
Sindaco R., Jeremcenko V. K. 2008. The Reptiles of the Western Palearctic. 1. Annotated Checklist and Distributional Atlas of the Turtles, Crocodiles, Amphisbaenians and Lizards of Europe, North Africa, Middle East and Central Asia. Latina, Italy: Edizioni Belvedere, 579 pp
Solovyeva E. N., Poyarkov N. A., Dunaev E. A., Duysebayeva T. N., Bannikova A. A. 2011. Molecular differentiation and taxonomy of the sunwatcher toad-headed agama species complex Phrynocephalus superspecies helioscopus (Pallas 1771) (Reptilia: Agamidae). Russ J Genet, 47: 842–856
Solovyeva E. N. 2013. Genetic variability and the phylogeny of the genus Phrynocephalus (Reptilia: Agamidae). Ph.D. Thesis. Lomonosov Moscow State University, Moscow, Russia (In Russian)
Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22: 2688–2690
Stamatakis A., Hoover P., Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web Servers. Syst Biol, 57: 758–771
Swofford D. L. 2003. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods), Version 4. Sunderland, MA: Sinauer
Szcerbak N. N. 1974. The Palearctic Desert Lizards (Eremias). Kiev: Naukova Dumka, 296 pp (In Russian)
Szcerbak N. N. 1975. Geographical variability and intraspecies taxonomy of Eremias velox Pall., 1771 (Reptilia, Sauria). Vestnik Zool, 6: 24–33 (In Russian with English abstract)
Szczerbak N. N. 2003. Guide to the Reptiles of the Eastern Palearctic. Malabar, Florida: Krieger Publishing Company, 260 pp
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol, 28: 2731–2739
Wang Y., Zhou L., Shi L. 2014. Scalation varaiton and subspecies classification status of rapid racerunner (Eremias velox) in Xinjiang. Sichuan J Zool, 33: 13–16 (In Chinese with English abstract)
Wiens J. J., Penkrot T. A. 2002. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Sys Biol, 51: 69–91
Xie W., Lewis P. O., Fan Y., Kuo L., Chen M.-H. 2011. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst Biol, 60: 150–160
Zhang Y., Stock M., Zhang P., Wang X., Zhou H., Qu L. 2008. Phylogeography of a widespread terrestrial vertebrate in a barely studied Palearctic region: green toads (Bufo viridis subgroups) indicate glacial refugia in eastern Central Asia. Genetica, 134: 353–365
Zhao K.-T., 1999. Lacertidae. In Zhao E.-M., Zhao K.-T., Zhou K.-Y. (Eds), Fauna 3 Sinica, Reptilia (Squamata: Lacertilia), Vol. 2. Beijing: Science Press, 219–242 (In Chinese)
Zhao Q., Liu H. X., Luo L. G., Ji X. 2011. Comparative population genetics and phylogeography of two lacertid lizards (Eremias argus and E. brenchleyi) from China. Mol Phylogenet Evol, 58: 478–491
Zink R. M. 2004. The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proc R Soc B, 271: 561–564
Zwickl D. J., Hillis D. M. 2002. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol 51, 588–598
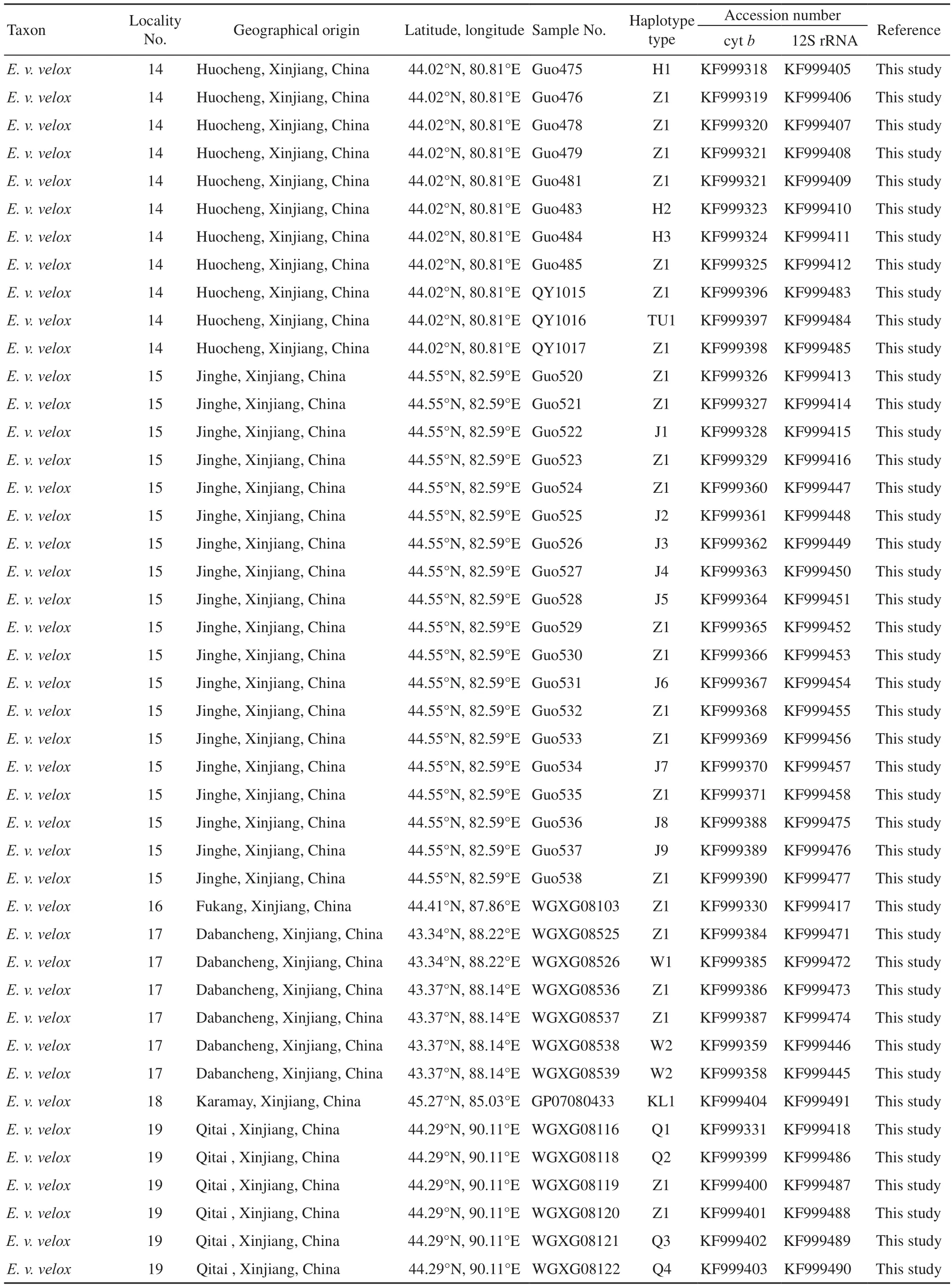
Appendix A List of analyzed specimens, their geographical origin and the GenBank accession number.
(Continued Appendix A)
(Continued Appendix A)
10.3724/SP.J.1245.2014.00012
*Corresponding author: Dr. Xianguang GUO, from Chengdu Institute of Biology, Chinese Academy of Sciences, with his research focusing on taxonomy, phylogeny and biogeography of reptiles.
E-mail: guoxg@cib.ac.cn
27 December 2013 Accepted: 1 March 2014
Asian Herpetological Research 2014, 5(1): 12–25
杂志排行

Asian Herpetological Research的其它文章
- Molecular Characterization and Expression Analysis of Matrix Metalloproteinase 3 in the Asian Yellow Pond Turtle Mauremys mutica
- Structure Organization of Urinary System in the Yellow Spotted Mountain Newts (Salamandridae: Neurergus microspilotus)
- A New Species of the Genus Tylototriton (Urodela: Salamandridae) from Northeastern Hunan Province, China
- Body Surface Area Prediction in Odorrana grahami
- Preliminary Insights into the Habitat Preferences of the Centralian Bandy Bandy (Vermicella vermiformis) (Squamata: Elapidae) in Central Australia
- Multilocus Phylogeny of Lycodon and the Taxonomic Revision of Oligodon multizonatum