乳化溶剂挥发法制备紫杉醇PLGA纳米粒及其体外评价
2014-06-15寇龙发高利芳李东坡
寇龙发,高利芳,姚 情,李东坡,孙 进
(沈阳药科大学 药学院,辽宁 沈阳 110016)
乳化溶剂挥发法制备紫杉醇PLGA纳米粒及其体外评价
寇龙发,高利芳,姚 情,李东坡,孙 进*
(沈阳药科大学 药学院,辽宁 沈阳 110016)
目的采用乳化溶剂挥发法制备紫杉醇的PLGA纳米粒,并对其进行体外评价。方法 基于单因素优化和正交设计优化考察了纳米粒制备的处方及工艺。对纳米粒的粒径、zeta电位、包封率和载药量、形态、稳定性及体外释放行为进行表征。 结果 采用动态光散射技术测定纳米粒粒径为(211.3±1.4) nm;测定zeta电位为(-4.42±0.70) mV;包封率和载药量分别为(91.4±3.2)%和(4.35±0.15)%;透射电镜结果显示纳米粒为球形,且分布均匀;体外稳定性良好,具有明显的缓释效果。结论 所制备的紫杉醇PLGA纳米粒粒径分布均一、包封率高、稳定性好,为进一步探索紫杉醇PLGA纳米粒的体内药动学奠定了实验基础。
药剂学;PLGA纳米粒;乳化溶剂挥发法;正交设计;紫杉醇
紫杉醇作为一种广谱抗肿瘤药物已广泛应用于临床,其主要通过诱导和促进微管蛋白的聚合,抑制微管解聚而使肿瘤细胞的有丝分裂终止,从而阻碍肿瘤细胞复制,使肿瘤细胞死亡[1]。紫杉醇属于BCS Ⅳ类药物,溶解性差,渗透性差,极大地影响了其临床应用;但近年来紫杉醇的制剂研究取得了较大进展。已经上市的有紫杉醇注射液(taxol)和紫杉醇白蛋白纳米粒(abraxane,capxol)。此外,如脂质体[2-3]、聚合物胶束[4-5]、聚合物纳米粒[6-7]及树枝状分子[8-9]等新的药物传递系统的出现,极大地改善了紫杉醇的体内外性质,为提高其临床应用奠定了坚实的基础。
纳米粒作为药物传递和控制释放的载体具有许多优点:(1)可有效保护药物,避免其降解,提高药物的体内稳定性;(2)可以控制药物释放,延长体内半衰期,提高生物利用度;(3)易实现靶向和定位;(4)体积微小,能够有效穿过血管壁到达肿瘤部位。在制备纳米载体的材料中,可生物降解的聚合物受到普遍重视并得到广泛的应用。聚乳酸-聚羟基乙酸共聚物[poly(lactic-co-glycolic acid), PLGA]是FDA批准的可用于注射剂的生物降解高分子聚合物。由于其具有良好的组织相容性和生物可降解性,优良的成囊性能和可调节的降解速率,PLGA已广泛应用于纳米药物传递系统的研究。
PLGA纳米粒的制备方法常用的主要有纳米沉淀法和乳化溶剂挥发法。纳米沉淀法制备的纳米粒所形成的粒径较小,但是重现性差,影响因素不易控。而乳化溶剂挥发法所制备的纳米粒粒径分布均匀,且产率较高。本文作者采用优化的乳化溶剂挥发法制备PLGA纳米粒,以经典抗癌药紫杉醇为模型药物,以期获得理想的粒径分布和较高的药物包封率,并对其体外稳定性及释药行为进行研究,为进一步研究紫杉醇PLGA纳米粒体内药动学行为奠定基础。
1 仪器和材料
AR1140电子分析天平(奥豪斯国际贸易有限公司),AG245电子分析天平(瑞士METTLER TOLEDO公司),H-600透射电镜(日本Hitachi 公司),Malvern Zeta Sizer(美国Malvern公司),LDZ5-2低速自动平衡离心机(北京医用离心机厂),PHS-3C酸度计(上海雷磁仪器厂),HITACHI高效液相色谱仪(包括UV Detector 5410、Column Oven 5310、Autosampler 5210、Pump 5110,日本日立公司),ECOSIL-C18色谱柱(150 mm×4.6 mm,5 μm, Thermo Fisher中国分公司),HZQ-C空气浴振荡机(哈尔滨东联电子技术开发有限公司),KQ-100超声波清洗器(昆山超声仪器有限公司)。
紫杉醇(paclitaxel,含量质量分数99.9%,重庆美联现代技术有限公司),聚乳酸-聚羟基乙酸共聚物(PLGA,乳酸与羟基乙酸物质的量比为50:50,相对分子质量为38 000,岱罡生物科技有限公司),聚乙烯醇(PVA,相对分子质量为20 000~30 000,含量质量分数88%,美国ACROS公司),聚氧乙烯蓖麻油(cremophor EL,巴斯夫中国有限公司),透析袋(截留相对分子质量:12 000~14 000,上海绿鸟科技发展有限公司),乙腈(色谱纯,天津康科德科技有限公司),水为重蒸水,其他试剂(分析纯,市售)。
2 方法
2.1 PLGA纳米粒的制备方法
采用乳化溶剂挥发法制备紫杉醇PLGA纳米粒。精密称取一定量的药物和PLGA,用1 mL有机相溶解,将其加入一定量的水相中,冰浴超声乳化分散一段时间,形成纳米乳,在室温下磁力低速搅拌一段时间,挥去有机溶剂,于13 000 r·min-1高速离心30 min收集纳米粒,以重蒸水反复洗涤3次后,冻干,制得紫杉醇PLGA纳米粒。
2.2 处方工艺优化
2.2.1 单因素实验
以纳米粒的制备方法为基础,以粒径为评价指标,采用单因素实验系统考察有机相的种类(二氯甲烷、氯仿、乙酸乙酯),表面活性剂的种类(PVA、Tween80、普朗尼克F68),表面活性剂的质量浓度( 5.0 g·L-1、10.0 g·L-1、20.0 g·L-1),油水相体积比(1:3、1:5、1:10),超声强度(50 W、100 W、200 W、300 W),超声时间(3 min、5 min、10 min)及挥去有机溶剂所用转速(500 r·min-1、1 000 r·min-1、1 500 r·min-1),确定最优处方工艺。
2.2.2 正交优化设计
在各单因素实验的基础上, 选取对紫杉醇PLGA纳米粒制备影响较大的4个因素: 乳化剂质量浓度、油水相体积比、超声强度、超声时间。每个因素选择3个水平, 按照L9(34) 正交表设计实验进行处方工艺优化。以粒径为评价指标,通过正交试验的直观分析优化处方及最佳制备工艺。因素水平设计表见表1。

表 1 正交设计中的因素和水平Table 1 Factors and levels of the orthogonal design
2.2.3 最优处方工艺验证
综合单因素实验和正交优化设计,确定最优处方和制备工艺。并按最优处方工艺进行3次重复验证试验,即分别制备3批紫杉醇PLGA纳米粒样品,测定其粒径。
2.3 纳米粒的表征
2.3.1 粒径和zeta电位测定
取PLGA纳米粒胶体溶液,以重蒸水稀释至适当浓度,于25 ℃下以动态光散射粒径分析仪测定纳米粒的粒径。用zeta电位仪测定纳米粒的zeta电位。
2.3.2 形态学考察(透射电子显微镜)
取PLGA纳米粒胶体溶液,以重蒸水稀释至适当浓度,取1、2滴纳米粒胶体溶液置于铜网上,用质量浓度10.0 g·L-1磷钨酸染色,室温晾干,于投射电子显微镜下观察纳米粒的形态并拍照。
2.3.3 载药量和包封率的测定
包封率和载药量是纳米粒质量评价的重要指标,采用微柱离心法测定纳米粒的载药量和包封率。按公式:包封率(EE)=m包/m药×100%计算载药纳米粒的包封率(EE)和公式:载药量(DL)= m包/m总×100%计算载药纳米粒的载药量(DL%)。式中,m包为纳米粒中包裹的药物质量(mg),m药为投入的总药质量(mg),m总为载药纳米粒的总质量(mg)。
2.4 PLGA纳米粒体外稳定性考察
将制成的纳米粒胶体溶液置于4 ℃冰箱中放置,于特定时间测定纳米粒的粒径变化。
2.5 载药纳米粒的体外释放实验
选取含有20.0 g·L-1的聚氧乙烯蓖麻油的pH值7.4的磷酸缓冲溶液(PBS)作为释放介质,考察载药纳米粒的释放特征。精密移取载药纳米粒胶体溶液2.0 mL于透析袋中,然后将密封的透析袋置于50 mL的释放介质中,平行操作3个样品,在37 ℃、100 次·min-1震荡的条件下进行体外释放试验。于特定时间内取出释放介质2 mL,并补充等量体积的新鲜介质。用HPLC法测定样品中紫杉醇的含量,计算药物的累积释放量和累积释放百分率。紫杉醇的测定波长为227 nm,流动相为乙腈-水(体积比50︰50),流速为1.0 mL·min-1。
3 结果和讨论
3.1 处方工艺优化单因素考察
在PLGA纳米粒的制备过程中,多种因素影响其粒径分布。主要考察了有机相种类、表面活性剂种类、表面活性剂质量浓度、油水相体积比、超声强度、超声时间、挥去有机溶剂所用转速对制备的纳米粒粒径的影响。
3.1.1 有机相种类的影响
乳化溶剂挥发法所用的有机相不能与水相混溶,才能在探头超声条件下很好地形成纳米乳。选择的二氯甲烷、氯仿、乙酸乙酯三种有机溶剂与水相混合超声后均能形成纳米乳,但是二氯甲烷的乳化效果最好,所以选用二氯甲烷为有机相。
3.1.2 表面活性剂种类的影响
分别采用10.0 g·L-1的普朗尼克F68、Tween80、PVA作为表面活性剂进行处方筛选,结果见图1A。制备过程中发现,以PVA为表面活性剂的样品会有淡蓝色乳光,而以Tween80和普朗尼克F68为表面活性剂的样品中没有乳光。由图1 A可知,以PVA作为表面活性剂可以得到相对较小的粒径及较小的PDI值。说明PVA作为表面活性剂可以有效降低PLGA纳米粒的粒径且控制其粒径分布在较窄范围内。
3.1.3 表面活性剂质量浓度的影响
分别采用不同质量浓度的PVA溶液作为表面活性剂,进行处方筛选,结果见图1B。由图1B可知,随着表面活性剂质量浓度的增大,PLGA纳米粒的粒径逐渐变大,选择10.0 g·L-1及其以下质量浓度可获得较小粒径。
3.1.4 油水相体积比的影响
分别以有机相与水相体积比为1:3、1:5、1:10进行处方筛选,结果见图1C。由图1C可知,随着水相比例的增大,PLGA纳米粒的粒径逐渐减小,且其比例增大到1:5时,粒径降低的速率减小。
3.1.5 超声强度的影响
分别以50、100、200、300 W的强度进行制备工艺考察,结果见图1D。由图1D可知,随着超声强度的增大,纳米粒粒径减小。当粒径下降到210 nm左右的时候不再下降。
3.1.6 超声时间的影响
分别以3、5、10 min的超声时间进行制备工艺的考察,结果见图1E。由图1E可知,随着超声时间的延长,纳米粒粒径减小,且超声时间超过5 min后,纳米粒粒径减小且速率变缓。
3.1.7 挥去有机溶剂所用转速的影响
分别以500、1 000、1 500 r·min-1的转速进行制备工艺的考察,结果见图1F。由图1F可知,转速对纳米粒粒径几乎不影响。实验操作中发现,不同的转速能够影响有机溶剂挥去的时间,转速越大,有机溶剂挥去越快。
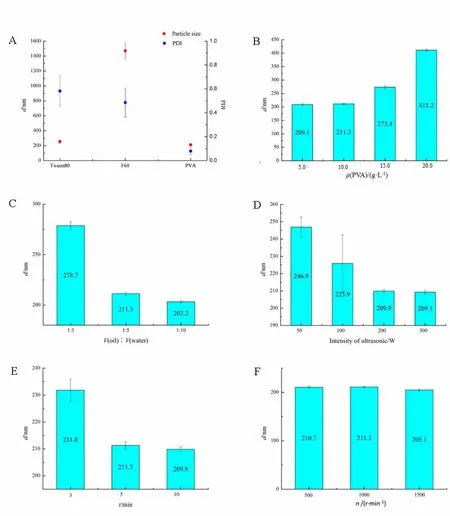
图1 纳米粒的处方工艺对粒径的影响Fig. 1 The effects of preparation and formulation of the nanoparticles on particle size
3.2 正交设计优化
在单因素分析的基础上,选取对纳米粒粒径影响较大的4个因素(表面活性剂质量浓度、油水相体积比、超声强度、超声时间)对其进行正交优化。正交实验设计结果见表2。

表 2 正交实验设计结果Table 2 The results of the orthoganol design
以粒径为指标进行直观分析,由极差R确定主次因素顺序为A>C>B>D,其中A因素K1>K3>K2,K2为最优水平;B因素K1>K2>K3,K3为最优水平;C因素K1>K2>K3,K3为最优水平;D因素K1>K2>K3,K3为最优水平。因此确定A2B3C3D3为最优处方,即表面活性剂选择质量浓度10.0 g·L-1PVA溶液,油水相体积比为1︰10,超声强度为300 W,超声时间为10 min。
3.3 粒径分布及zeta电位
以最优处方工艺制备PTX-PLGA-NPs(紫杉醇PLGA纳米粒),动态光散射粒径测定仪测定结果为PTX-PLGA-NPs的平均粒径为(211.3±1.4) nm(图2),分布均匀;zeta电位为(-4.42±0.70) mV。由于使用的PLGA的羧基端连有甲基封端,因此制备的纳米粒所显示电负性并不明显,而呈相对中性。

图 2 紫杉醇PLGA纳米粒的粒径分布Fig. 2 The size distribution of paclitaxel loaded PLGA nanoparticles
3.4 形态学考察(TEM)
透射电镜结果(图3、4)显示,制得的紫杉醇PLGA纳米粒呈球形,表面圆整光滑,粒子之间无黏连现象,分散性良好,且粒径分布较为均一。与动态光散射结果相比,此粒径偏小。原因是动态光散射粒径仪测定时样品处于水环境中,离子表面较为伸展,且有表面活性剂PVA的存在,在纳米粒周围形成一定范围的水化层;而拍摄TEM时样品制备需要干燥,导致粒子会发生轻微皱缩。图3中PLGA纳米粒未经过洗去表面活性剂的步骤,因此周围有一层由PVA形成的晕;而图4中的PLGA纳米粒经过多次洗去表面活性剂的步骤,纳米粒表面的环状晕减少。

图 3 紫杉醇PLGA纳米粒的透射电镜照片(未洗去表面活性剂)Fig. 3 TEM image of the paclitaxel-loaded PLGA nanoparticles (without washing)

图 4 紫杉醇PLGA纳米粒的透射电镜照片(洗去表面活性剂)Fig. 4 TEM image of the paclitaxel-loaded PLGA nanoparticles (washed)
3.5 包封率和载药量的测定
采用本研究优化后的处方工艺(有机相为二氯甲烷,水相为10.0 g·L-1PVA溶液,油水相体积比为1:10,超声强度为300 W,超声时间10 min)重复制备紫杉醇PLGA纳米粒3批,以微柱离心法测定其包封率和载药量。测得纳米粒的平均包封率为(91.4±3.2)%,平均载药量为(4.35±0.15)%,且制备工艺的重复性良好。
3.6 PLGA纳米粒的体外稳定性试验
由PLGA纳米粒的稳定性实验结果(图5)可知,4 ℃条件下,PLGA纳米粒在15 d内粒径基本不发生变化,由PDI值可看出粒径分布变化不大。由此得出,本研究所制备的PLGA纳米粒稳定性良好。
3.7 紫杉醇PLGA纳米粒的体外释放试验
紫杉醇在水中的溶解度低,当用普通溶出介质时,溶出液中只有痕量的药物,文献中有报道采用1.0 g·L-1Tween 80[10-11]、10.0 g·L-1Tween 80[12]、20.0 g·L-1Tween 80[13]等作为紫杉醇体外释放的增溶剂。但是经实验验证后发现,释放过程中会有沉淀在透析袋中产生,增溶效果并不理想;释放介质中加入20.0 g·L-1的cremophor EL[14]可以有效增加紫杉醇在释放介质中的溶解度,使其达到漏槽条件,故本实验采用20.0 g·L-1cremophor EL作为增溶剂。使用含有20.0 g·L-1cremophor EL的pH值7.4的PBS作为释放介质研究PLGA纳米粒中紫杉醇的释放特征(图6)。

图 5 P LGA纳米粒在4 ℃的稳定性Fig. 5 S tability of PLGA n anoparticles at 4 ℃

图 6 紫杉醇PLGA纳米粒的体外释放行为(n=3)Fig. 6 Release behavior of the paclitaxel-loadedPLGA nanoparticles (n=3)
从结果可以看出,PLGA纳米粒可以控制紫杉醇缓慢释放,并且没有明显的突释现象;72 h以内的释放接近于零级释放,并且在第72小时累积释放度达到70%以上;结果显示样品累积释放度在96 h后出现不同程度的下降,可能是因为有部分的紫杉醇降解。
4 结论
以PLGA为载体材料,以乳化溶剂挥发法制备了紫杉醇的生物可降解给药系统PTX-PLGA-NPs。为获得具有理想粒径的PTX-PLGA-NPs,本文对纳米粒的处方及制备工艺进行了筛选优化。在单因素实验的基础上,采用正交设计优化处方,得到最优处方。以最优处方制得的PTX-PLGA-NPs平均粒径为(211.3±1.4) nm,zeta电位为(-4.42±0.70) mV,平均包封率为(91.4±3.2)%,平均载药量为(4.35±0.15)%。对其进行形态学、体外稳定性、体外释药特征分析,PTX-PLGA-NPS表面圆整光滑,理化性质稳定,具有良好的药物缓释性能,为抗肿瘤缓释制剂的开发提供了实验基础。在本研究基础上,后续工作将进一步探索PTX-PLGA-NPs的体内药动学规律,为其临床应用提供相应的实验基础。
[1] KUMAR N. Taxol-induced polymerization of purified tubulin. Mechanism of action[J]. J Biol Chem, 1981, 256: 10435-10441.
[2] SEZGIN-BAYINDIR Z, ONAY-BESIKCI A, VURAL N, et al. Niosomes encapsulating paclitaxel for oral bioavailability enhancement: preparation, characterization, pharmacokinetics and biodistribution[J]. J Microencapsul, 2013, 30: 796-804.
[3] BAYINDIR Z S, YUKSEL N. Characterization of niosomes prepared with various nonionic surfactants for paclitaxel oraldelivery[J]. J Pharm Sci, 2010, 99: 2049-2060.
[4] DAHMANI F Z, YANG Hui, ZHOU Jian-ping, et al. Enhanced oral bioavailability of paclitaxel in pluronic/LHR mixed polymeric micelles: preparation, in vitro and in vivo evaluation[J]. Eur J Pharm Sci, 2012, 47: 179-189.
[5] LI Yi-mu, BI Yi-ling, XI Yan-wei, et al. Enhancement on oral absorption of paclitaxel by multifunctional pluronic micelles[J]. J Drug Target, 2013, 21: 188-199.
[6] ZHAO Ling-yun, FENG Si-shen. Enhanced oral bioavailability of paclitaxel formulated in vitamin E-TPGS emulsified nanoparticles of biodegradable polymers: In vitro and in vivo studies[J]. J Pharm Sci, 2010, 99: 3552-3560.
[7] MU Li, FENG Si-shen. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS[J]. J Controlled Release, 2003, 86: 33-48.
[8] OOYA T, LEE J, PARK K. Hydrotropic dendrimers of generations 4 and 5: synthesis, characterization, and hydrotropic solubilization of paclitaxel[J]. Bioconjug Chem, 2004, 15: 1221-1229.
[9] MAJOROS I J, MYC A, THOMAS T, et al. PAMAM dendrimer-based multifunctional conjugate for cancer therapy: synthesis, characterization, and functionality[J]. Biomacromolecules, 2006, 7: 572-579.
[10] FENG Si-shen, ZENG Wu-tao, LIM Yean-teng, et al. Vitamin E TPGS-emulsified poly (lactic-co-glycolic acid) nanoparticles for cardiovascular restenosis treatment[J]. Nanomedicine, 2007, 2(3): 333-344.
[11] KIM B S, KIM C S, LEE K M. The intracellular uptake ability of chitosan-coated Poly (D, L-lactideco-glycolide) nanoparticles[J]. Arch Pharm Res, 2008, 31: 1050-1054.
[12] XIE Jing-wei, WANG Chi-hwa. Self-assembled biodegradable nanoparticles developed by direct dialysis for the delivery of paclitaxel[J]. Pharmaceutical Research, 2005, 22: 2079-2090.
[13] KOLLIPARA S, BENDE G, MOVVA S, et al. Application of rotatable central composite design in the preparation and optimization of poly (lactic-co-glycolic acid) nanoparticles for controlled delivery of paclitaxel[J]. Drug Dev Ind Pharm, 2010, 36: 1377-1387.
[14] LIAN He, ZHANG Tian-hong, SUN Jin, et al. Enhanced oral delivery of paclitaxel using acetylcysteine functionalized chitosan-vitamin E succinate nanomicelles based on a mucus bioadhesion and penetration mechanism[J]. Molecular Pharmaceutics, 2013, 10: 3447-3458.
Preparation and in vitroevaluation of paclitaxelloaded PLGA nanoparticles by emulsion-solvent evaporation method
KOU Long-fa, GAO Li-fang, YAO Qing, LI Dong-po, SUN Jin*
(School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China)
ObjectiveTo evaluate the paclitaxel (PTX) loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles prepared by emulsion-solvent evaporation method. Methods The preparation and formulation of the nanoparticles were optimized with single factor tests and an orthogonal design. ThePTX-PLGA nanoparticles were characterized with respect to particle size, zeta potential, entrapment efficiency, drug loading, shape, stability and release behavior. Results The mean particle size measured by dynamic light scattering (DLS) was (211.3±1.4) nm, and the zeta potential was (-4.42±0.70) mV. The entrapment efficiency and the drug loading of PTX-NPs were (91.4±3.2)% and (4.35±0.15)%, respectively. The images of TEM show that the nanoparticles were spherical and uniform. In vitro, the PTX-PLGA nanoparticles present good stability and sustained-release behavior. Conclusions The resultant nanoparticles have uniform particle size, high drug loading and good stability. These results are the experimental basis in vitro for the further in vivo pharmacokinetic research of PTX-PLGA nanoparticles.
Pharmaceutics; PLGA nanoparticles; emulsion-solvent evaporation method; orthogonal design; paclitaxel
R 94
A
(2014)02–0033–10
(本篇责任编辑:马丽丽)
2013–12–31
寇龙发(1988-),男(汉族),河南偃师人,硕士研究生,E-mail klf.666666@163.com;*通讯作者:孙进(1975-),男(汉族),安徽金寨人,教授,博士,主要从事药剂学研究,Tel. 024-23986325,E-mail sunjin0529@aliyun.com。
