Ag掺杂AlN半导体光电性质的第一性原理研究
2014-06-09邓军权毋志民王爱玲赵若禺胡爱元
邓军权, 毋志民, 王爱玲, 赵若禺, 胡爱元
(重庆师范大学物理与电子工程学院,重庆 401331)
Ag掺杂AlN半导体光电性质的第一性原理研究
邓军权, 毋志民*, 王爱玲, 赵若禺, 胡爱元
(重庆师范大学物理与电子工程学院,重庆 401331)
采用基于密度泛函理论的第一性原理平面波超软赝势法,对Ag掺杂AlN 32原子超晶胞体系进行几何结构优化,计算并分析体系的电子结构、磁性和光学性质.结果表明:Ag掺杂后,Ag4d态电子与其近邻的N2p态电子发生杂化,引入杂质带形成受主能级,实现p型掺杂,使体系的导电能力增强,同时表现出金属性和弱磁性,其净磁矩为1.38 μв.掺杂形成的N-Ag键电荷集居数较小,表现出强的离子键性质.掺杂后体系的介电函数虚部和光吸收谱在低能区出现新的峰值,同时复折射率函数在低能区发生变化,吸收边向低能方向延展,体系对长波吸收加强,能量损失明显减小.
Ag掺杂AlN;电子结构;铁磁性;光学性质;第一性原理
0 引言
III-V族化合物氮化铝(AlN)是一种新型的直接带隙宽禁带半导体材料[1],禁带宽度(Eg)为6.2 eV[2],常温常压下的稳定相是六方纤锌矿结构[3],在许多方面表现出非常独特的物理化学性能,具有广阔的应用前景.因具有较宽的禁带、低的介电常数和高机械强度及与硅相近的热膨胀系数,使其成为微电子器件中理想的基底材料[4].而AlN还具有良好的压电性质和较高的机电耦合系数,是GHz级声表面波装置的优选压电材料[5].同时,良好的热稳定性、化学稳定性和低介电损耗,可使AlN用于大规模集成电路和大功率器件散热材料[6].并且AlN还是重要的蓝光、紫外发光材料[7],也是目前制作短波长的紫外发光二极管固体光源的重要材料[5,8].此外AlN还具有无毒性,无环境污染和生产成本相对低廉等优点,是一种环保材料,在水和空气净化、消毒等方面也具有潜在应用价值[9-10].因而AlN成为近年来半导体领域的研究热点之一.
目前人们对AlN的研究主要集中在两个方面.一方面是致力于获得n型或p型半导体材料,其方法大都是向AlN中引入杂质离子.如用Si掺杂AlN便可实现强电导性能的n型材料[11].Zn、Cd掺杂AlN可以为体系提供较多的空穴态,实现p型材料[10,12].Mg掺杂AlN不仅实现了p型材料,还以此研制成了发光波长为210 nm的发光二极管[9].另一方面则是致力于得到同时兼具电荷属性和自旋特性的稀磁半导体(DMS)材料[13-14],其方法一般是引入磁性过渡族离子.如Mn、Fe掺杂AlN可实现100%的自旋极化载流子注入,半金属能隙达到0.727 eV,体系净磁矩达5 μв[15].12.5%的Cr掺杂AlN的半金属能隙可达1.09 eV[16].然而由于用磁性过渡金属掺杂体系的磁沉积问题和铁磁性机理不易解释清楚,于是有研究人员尝试用非磁性离子进行掺杂,如Cu掺杂AlN得到的净磁矩为2 μв,磁性的产生被解释为源于p-d电子杂化[17-18].
当今无论是在制备p、n型AlN半导体材料还是在获取AlN稀磁半导体材料上,所面临的共同难题是掺杂效率太低,这主要是掺入杂质的固溶度较低和离化能较高造成的[19],从而制约了AlN的发展.进一步探究性质更好的受主成分是解决这一难题的渠道之一.已有研究表明,Ag具有电导率高,化学性质稳定等优点,且为非磁性离子,在掺杂ZnO中能得到光学质量较好的p型材料,同时还能增加声子反射,降低热导率[20-21].第一性原理计算已广泛应用于材料的性质研究[22-23].本文采用基于密度泛函理论(DFT)的平面波超软赝势法计算分析Ag掺杂AlN前后体系的电子结构、磁电特性和光学性质,为实验研究提供有意义的参考.
1 模型结构与计算方法
1.1 模型构建
理想的AlN为六方纤锌矿结构,属于P63mc空间群,对称性为C6V-4,晶格参数为a=b=0.311 2 nm,c=0.498 2 nm,其中c/a为1.600 8[24].其晶胞由Al的六角密堆积与N的六角密堆积反向套构而成,比理想的六角密堆积结构的1.633小.计算基于超晶胞模型,取2×2×2(32原子)超晶胞体系,每个超晶胞包含16个Al原子和16个N原子.掺杂时,由一个Ag原子替代体系中的一个Al原子实现掺杂,掺杂浓度为6.25%.超晶胞模型如图1所示.

图1 32原子体系超晶胞结构图Fig.1 Supercell structure of AlN with 32 atoms
1.2 计算方法
计算由基于密度泛函理论的从头算量子力学程序CASTEP[25]完成.计算中采用周期性边界条件,利用广义梯度近似(GGA)中的PBE[26]近似处理电子间的交换关联能.电子波函数采用平面波基超软赝势法(PWP)[27]描述离子实与价电子间的相互作用,选取Al,N,Ag的价电子组态分别为Al:3s23p1,N:2s22p3,Ag:4d105s1.在倒易的K空间中,计算选取的平面波截断能(Ecut)为500 eV.体系总能和电荷密度在对布里渊区(Brillouin)的积分计算采用Monkhorst-Park[28]方案,对超晶胞体系选取K网格点为6×6×4,其自洽收敛精度设为5.0×10-7eV·atom-1.结构优化中采用BFGS[29]算法优化,其原子间相互作用力收敛标准设为0.01 eV,单原子能量收敛标准5.0×10-6eV·atom-1,晶体内应力收敛标准为0.02 GPa,原子的最大位移收敛标准为5.0×10-5nm.晶胞结构优化后,各项参数均优于收敛标准.
2 结果与讨论
2.1 纯AlN的电子结构
为与掺杂体系性质比较,先计算纯AlN体系的性质.为了让计算结果更加精确,计算选取的平面波截断能(Ecut)由400 eV增至500 eV,K网格点由4×4×2增至6×6×4,在保障体系收敛的同时逐渐提高精度.计算结果由表1所示,计算所得最佳晶格参数a=b=0.3112 nm,c=0.4921 nm与实验值[21](a=b=0.3112 nm,c=0.498 2 nm)符合较好.以后的计算均选取Ecut为500 eV,K网格点为6×6×4.计算所得的带隙值为Eg=4.676 eV,比Miwa[30],聂招秀等[31]人的计算结果(4.09 eV,4.10 eV)更接近实验值,但与实验值[2](6.2 eV)比较仍是低估的,这是因为计算中采用的DFT为基态理论,而能系属于激发态,因此计算结果偏低,但是并不影响对AlN及其掺杂体系电子结构和性质的理论分析.
图2为AlN超晶胞的能带结构和态密度.由图可以看出,AlN为直接带隙半导体,导带底和价带顶位于布里渊区的高对称点G点处.能带由-14.95 eV~-12.21 eV的下价带、-5.66 eV ~0 eV的上价带和4.68 eV~7.74 eV的导带构成.图2(a)中嵌入的小图为能带在费米能级处的局部放大图,可以看出,价带顶出现三个子带,分别是简并的重空穴、轻空穴和自旋-轨道耦合所分裂出的劈裂带,这和其他III-V族氮化物的能带结构相似[10].结合态密度图2(b)可以看出,价带主要由N的2s 和2p态构成,而导带主要由Al的3p态构成,其中价带的N2s,2p态电子态密度局域性较强,Al3s,Al3p态电子态密度则相对较弥散,整体上表现出一定的离子键性质.由总的态密度可以看出体系自旋向上和自旋向下的能带对称,没有产生自旋劈裂现象,体系没有净磁矩.
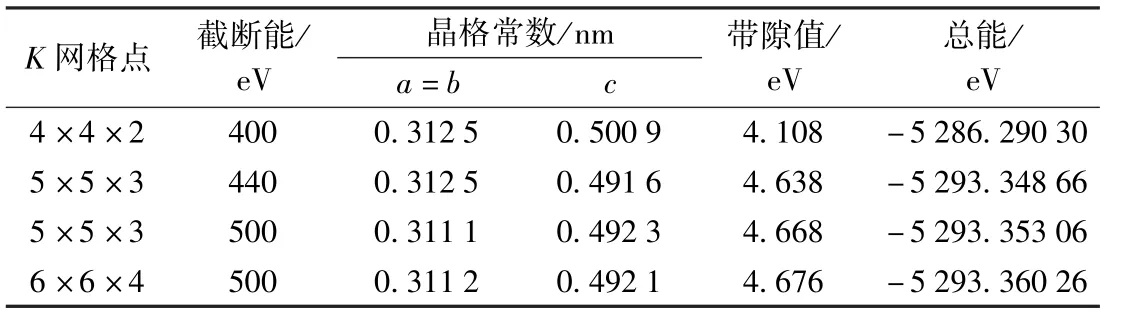
表1 不同截断能和K格点下计算的AlN的晶格常数、带隙值、总能Table 1 Calculated lattice constant,band gap,and total energy of AlN with different cut-off energy and K-points

图2 AlN超胞的能带结构(a)和态密度(b)Fig.2 Band structure(a)and density of states(b)of AlN supercell
2.2 Ag掺杂AlN的电子结构
表2给出了Ag掺杂AlN体系结构优化后的晶格常数、总能和带隙值,与纯AlN对比可知,Ag掺杂后体系的晶格常数略有增大,这主要是因为Ag离子半径比Al离子大的缘故.其次是体系的带隙值明显减小,表明Ag的掺入增强了体系的电导能力.掺杂体系的稳定性可根据体系的结合能Δ[29]来判断,若Δ为负值,表明体系较稳定,若Δ为正值,则体系不稳定,且Δ越大体系越不稳定,实验上要掺入杂质就越困难.Δ=E(AlAgN)-E(AlN)+μ(Al)-μ(Ag),其中E表示计算的体系的总能,μ为化学势,μ(Ag)=-1 027.755 59 eV,μ(Al)=-56.417 98 eV.计算得Δ=6.859 40 eV,表明Ag掺入之后体系的稳定性较纯AlN有所降低.
图3为Ag掺杂AlN在费米能级附近的自旋极化能带图.可以看出,自旋向上和自旋向下的能带带隙依然存在,这表明Ag的掺入并没有破坏AlN整体上的半导体性质.与其他过渡族金属离子[15,18]掺杂AlN相同的是,掺杂之后在价带顶上方都引入了自旋极化杂质带.而不同的是,Ag掺入之后,体系能带结构无论自旋向上还是自旋向下,价带顶均跨过了费米能级,表现出了金属性质,明显提高了体系的导电性能.且Ag离子的掺入在费米能级上方引入了受主能级,即在费米能级附近引入了空穴载流子,表明Ag掺杂AlN为p型掺杂.由跨过费米能级的子带可以看出引入的空穴具有较大的有效质量,属重空穴,同时也引入了一定量的有效质量较小的轻空穴[10,33].由自旋向上和自旋向下的能带图对比可以看出,引入的杂质带在费米能级附近发生了劈裂现象,但劈裂程度不大,表明掺杂体系存在一定的净磁矩.

表2 Ag掺杂前后AlN的晶格常数、总能和带隙值Table 2 Lattice constant,total energy,and band gap of AlN before and after Ag doped

图3 Ag掺杂AlN的自旋向上能带图(a)和自旋向下能带图(b)Fig.3 Spin polarized band structures of Ag-doped AlN:(a)spin up,(b)spin down
结合图4的Ag掺杂AlN体系的态密度图可以看出,在费米能级附近跨过费米能级的子带,主要是由N 的2p态电子和Ag的4d态电子构成.由图4(a)可知,在费米能级附近N2p态电子和Ag4d态电子出现明显的态密度交迭,尤其是在-0.5 eV~1.0 eV之间,N2p与Ag4d态密度峰几乎完全重合,表明Ag4d电子与其最近邻的N2p电子发生明显杂化,使掺杂体系能带跨过费米能级,形成深受主能级,并表现出一定的金属性质.对费米能级以下的自旋向上和自旋向下态密度分别进行积分计算,得到自旋向上的电子数多余自旋向下的电子数,体系净磁矩为1.38 μв.其中Ag离子贡献0.26 μв,Ag离子周围的四个N原子贡献1.04 μв,其余原子贡献甚微.Ag掺入AlN后在价带顶引入了较多的空穴态,因为空穴的存在使费米能级远离价带顶而移入价带中,从而使费米能级处有较高的态密度.根据Peng[34]等人的理论,在离子性较强的含氮化合物中,氮元素有很强的自旋交换作用,同时空穴的存在使得价带顶附近有很高的态密度,故很容易使含氮化合物产生磁性,所以体系主要磁矩贡献为Ag四周的N原子.

图4 Ag掺杂AlN中Ag、N的分波态密度(a);Al的分波态密度(b);总的态密度(c)Fig.4 Density of states of Ag-doped AlN:(a)partial DOS of Ag and N,(b)partial DOS of Al,(c)total DOS
表3是AlN掺杂体系的键长和电荷布居重叠数.由表3可知AlN沿a-b轴方向的键长比沿c轴方向的键长短,成键强度与共价性较强.当Ag离子掺入后,因其电负性比Al小,与N所成的N-Ag键强度很弱,表现出反键和较强的离子键性质.掺杂体系的N-Al键总体上还是表现出共价性,其中靠近Ag离子沿a-b方向的成键减弱,沿c方向的成键增强.而远离Ag离子沿a-b方向的成键增强,沿c方向的成键减弱.这和前面计算体系的稳定性降低相吻合,而N-Ag成强的离子键很容易使掺杂体系产生磁性,这也和体系产生净磁矩相互印证.
2.3 Ag掺杂AlN的光学性质
对材料光学性质的研究,是分析研究材料电子结构等各种相关物理性质的有效技术手段,本文计算并分析了掺杂前后体系的介电函数、复折射率函数、光吸收谱和能量损失谱.根据半导体光学性质,在线性响度范围内半导体的宏观光学性质能够用复介电函数ε(ω)=εr(ω)+iεi(ω)来描述,其中,εr=n2(ω)+k2(ω),εi=2nk.根据直接跃迁定义和克喇末-克朗尼格(Krames-Kronig)色散关系可推得晶体的介电函数实部、虚部、反射率、吸收系数、复折射率等[35].
纯AlN和Ag掺杂AlN的介电函数虚部如图5(a)所示.能级间的跃迁产生了其光谱,可由能带结构和态密度来解释其介电峰的来源[36].从图5(a)可以看出,AlN主要有对应光子能量为E1=7.75 eV,E2=11.15 eV的两个特征峰,其中E1介电峰较强,E2介电峰较弱.对应到态密度图2(b)可知,E1介电峰对应着体系的直接跃迁阈,主要是价带N2p态电子向导带Al3p态跃迁的结果.这与体系带隙存在偏差的一个原因是电子跃迁吸收能量应考虑跃迁过程中的弛豫效应,而不是简单的两个能级差[37].Ag掺杂后,一个明显的现象是体系在0.67 eV左右新增了一个很高的介电峰E3,且之前的两个介电峰E1、E2峰值有一定的减弱.对比态密度图4(a)可知,在能量为0.67 eV左右对应的是价带电子向杂质带的跃迁结果,主要是N2p态与Ag4d态跃迁的结果.掺杂后E1、E2峰向低能方向略有偏移,主要是因为Ag的掺入使带隙变窄的缘故,而峰值变弱则是由于掺杂引入杂质带后,使各能级间的跃迁几率减小.
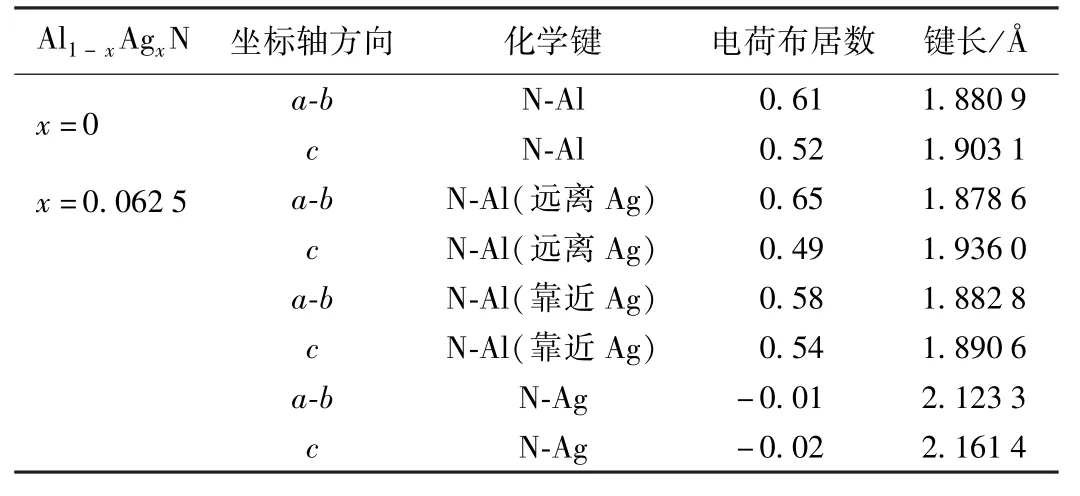
表3 Ag掺杂前后AlN的电荷布居数与键长Table 3 Charge population and bond lengths of AlN before and after Ag doped

图5 Ag掺杂前后AlN的介电函数虚部(a)和复折射率函数(b)Fig.5 Imaginary part of dielectric function(a)and complex refractive index function(b)of AlN before and after Ag doped
由图5(b)的复折射率函数可以看出,纯AlN在E<4.0 eV的低能区虚部n(εi)为0,而实部趋于常数.在E>13.0 eV的高能区虚部n(εi)的值为0,实部n(εr)的值也逐渐趋于常数,表明AlN体系对过低频和过高频的电磁波的吸收均较弱,吸收仅限定在一定的频率范围内.比较Ag掺杂后的复折射率函数可以看出,在高能区图形形状基本一致,但在低能区实部和虚部有明显变化,不再趋于常数,主要是因为Ag掺杂后,电磁波将通过不同的介质,造成折射率函数发生变化,增大了体系对低频电磁波的吸收.
图6(a)是Ag掺杂AlN前后体系的光吸收谱图.纯AlN吸收边能量对应的是4.0 eV和13.0 eV,与前面计算的复折射率函数相符.吸收主峰在8 eV左右,这与体系的直接跃迁阈对应.Ag掺杂之后在低能区出现了一个新的吸收峰,正好与受主能级电子的跃迁、介电函数特征峰E3和复折射率函数在低能区的变化对应.吸收峰减弱也和掺杂后体系引入受主能级使各能级间的跃迁几率变小相符合.由图6(b)的能量损失谱可以看出,纯净的AlN的损失峰位于13.8 eV左右,掺杂后损失峰向低能方向略微偏移,且能量损失明显减小,大约只有掺杂前的16%.
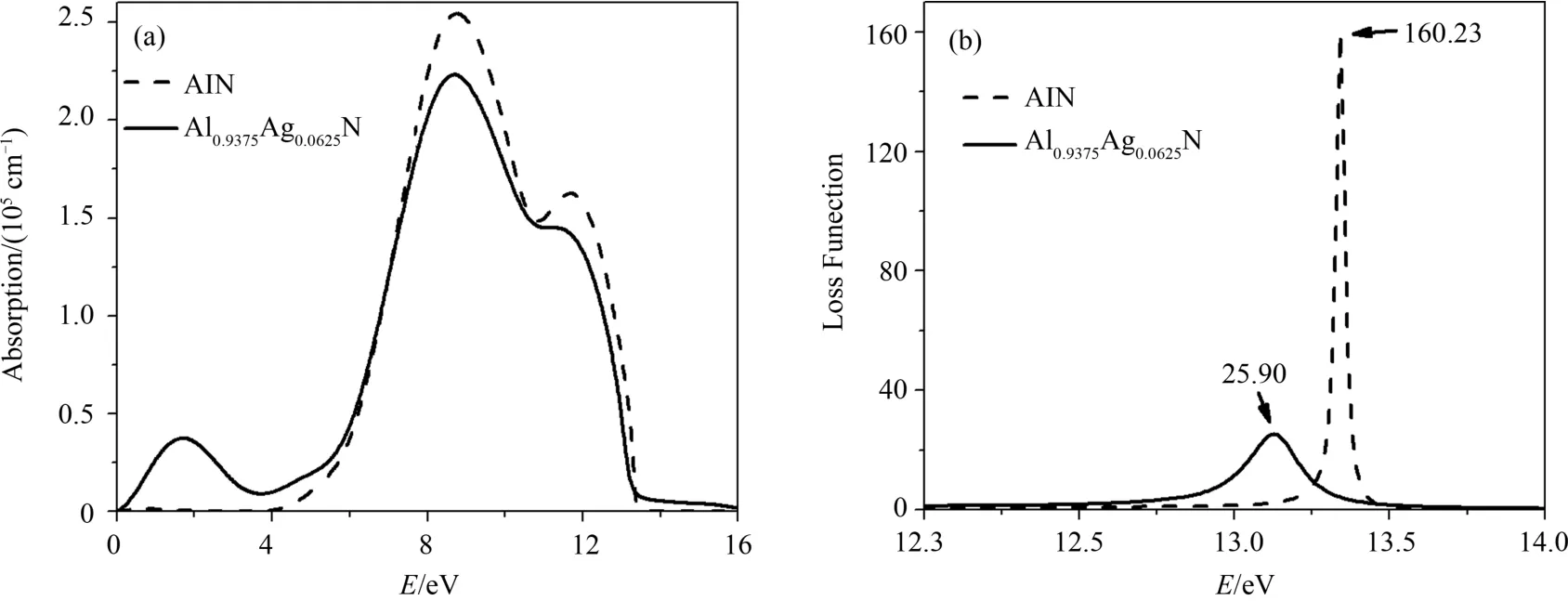
图6 Ag掺杂前后AlN的光吸收谱(a)和能量损失谱(b)Fig.6 Optical absorption spectra(a)and energy loss spectra(b)of AlN before and after Ag doped
3 结论
采用基于密度泛函理论的第一性原理平面波超软赝势法,对理想纤锌矿AlN及Ag掺AlN的超晶胞结构进行了几何优化,计算并分析了它们的电子结构、磁电性质和光学性质.结果表明:Ag掺杂AlN后,Ag4d电子与其近邻的N2p电子发生杂化,引入的杂质带无论自旋向上还是自旋向下均跨过费米能级,形成深受主能级,且使体系表现出一定的金属性,增大了体系的导电能力,表明Ag掺杂AlN是良好的p型掺杂剂.同时体系因Ag和N的自旋交换作用而产生1.38 μв的净磁矩.掺杂形成的N-Ag键电荷集居数很小,成键很弱,表现为强的离子键性质,而Al-N键受掺杂影响各有不同程度变化,使体系稳定性降低.掺杂后体系介电函数虚部和光吸收谱在低能区出现新的峰值,吸收边向低能方向延展,复折射率函数在低能区也发生明显变化,增强了体系对低频电磁波的吸收.Ag掺杂后体系能量损失明显减小,仅为之前的16%.
[1]Li J,Nam K B,Nakarmi M L,et al.Band structure and fundamental optical transitions in wurtzite AlN[J].Appl Phys Lett,2003,83(25):5163-5165.
[2]Taniysu Y,Kasu M,Makimoto T.Electrical conduction properties of n-type Si-doped AlN with high electron mobility(>cm 1002 V-1s-1)[J].Appl Phys Lett,2004,85(20):4672-4674.
[3]耶红刚,陈光德,竹有章,等.六方AlN本征缺陷的第一性原理研究[J].物理学报,2007,56(9):5376-5381.
[4]朱军山,徐岳生,郭宝平,等.Si(111)衬底上生长的GaN的形貌与AlN缓冲层生长温度的关系[J].半导体学报,2005,26(8):1577-1582.
[5]黄继颇,王连卫,林成鲁.性能优异的多功能宽禁带半导体AlN薄膜[J].功能材料,1999,30(2):141.
[6]周继承,石之杰.AlN电子薄膜材料的研究进展[J].材料导报,2007,21(5):14-17.
[7]Han J,Crawford M H,Shui R J,et al.AlGaN/GaN quantum well ultraviolet light emitting diodes[J].Appl Phys Lett,1998,73(12):1688-1690.
[8]Schubert E F,Kim J K.Solid-state light sources getting smart[J].Science,2005,308(5726):1274-1278.
[9]Taniysu Y,Kasu M,Makimoto T.An aluminium nitride light-emitting diode with a wavelength of 210 nanometres[J]. Nature,2006,441(7091):325-328.
[10]董玉成,郭志友,毕艳军,等.Zn,Cd掺杂AlN电子结构的第一性原理计算[J].发光学报,2009,30(3):314-320.
[11]Taniysu Y,Kasu M,Kobayashi N.Intentional control of n-type conduction for Si-doped AlN and AlxGa1-xN(0.42≤x<1)[J].Appl Phys Lett,81(7):1255-1257.
[12]Nepal N,Nakarmi M L,Jang H U,et al.Growth and photoluminescence studies of Zn-doped AlN epilayers[J].Appl Phys Lett,2006,89(19):192111.
[13]Ohno H.Making nonmagnetic semiconductors ferromagnetic[J].Science,1998,281(5379):951-956.
[14]Song Dewang,Niu Yuan,Xiao Liou,et al.First-principles study of structural,electronic,and magnetic properties of Mndoped ZnS(111)surfaces[J].Chinese J Comput Phys,2013,30(5):783-790.
[15]Doumi B,Tadjer A,Dahmane F,et al.Investigations of structural,electronic,and half-metallic ferromagnetic properties in (Al,Ga,In)1-xMxN(M=Fe,Mn)diluted magnetic semiconductors[J].J Supercond Nov Mag,2013,26(3):515-525.
[16]樊玉勤,何阿玲.基于第一性原理的Mn-AlN和Cr-AlN的半金属性质[J].物理化学学报,2010,26(10):2801-2806.
[17]Ji X H,Lau S P,Yu S F,et al.Ferromagnetic Cu-doped AlN nanorods[J].Nanotechnology,2007,18(10):105601-105604.
[18]林竹,郭志友,毕艳军,等.Cu掺杂的AlN铁磁性和光学性质的第一性原理研究[J].物理学报,2009,58(3):1917-07.
[19]张勇.掺杂AlN的理论与实验研究[D].武汉:华中科技大学,2008.
[20]吴子华,谢华清,曾庆峰.Ag-ZnO纳米复合热电材料的制备及其性能研究[J].物理学报,2013,62(9):97301-97301.
[21]王经纬,边继明,孙景昌,等.Ag掺杂p型ZnO薄膜及其光电性能研究[J].物理学报,2008,57(8):5212-5216.
[22]Liu Xiankun,Zheng Zhou,Lan Xiaohua,et al.First-principles study of structure,elastic and thermodynamic properties of ZrV2[J].Chinese J Comput Phys,2013,30(2):256-264.
[23]Peng Qiang,Yang Xiaoxi,Ding Jing,et al.Thermodynamic properties of cubic KNO2under atmospheric pressures:Density functional study[J].Chinese J Comput Phys,2013,30(2):271-276.
[24]Ishihara M,Li S J,Yumoto H,et al.Control of preferential orientation of AlN films prepared by the reactive sputtering method [J].Thin Solid Films,1998,316(1):152-157.
[25]Segall M D,Lindan P J D,Probert M J,et al.First-principles simulation:Ideas,illustrations and the CASTEP code[J]. Journal of Physics:Condensed Matter,2002,14(11):2717.
[26]Perdew J P,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Physical Review Letters,1996,77 (18):3865.
[27]Vanderbilt D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Physical Review B,1990,41 (11):7892.
[28]Monkhorst H J,Pack J D.Special points for Brillouin-zone integrations[J].Physical Review B,1976,13(12):5188-5192.
[29]Fischer T H,Almlof J.General methods for geometry and wave function optimization[J].The Journal of Physical Chemistry,1992,96(24):9768-9774.
[30]Miwa K,Fulumoto A.First-principles calculation of the structural,electronic,and vibrational properties of gallium nitride and aluminum nitride[J].Phys Rev B,1993,48(11):7897–7902.
[31]聂招秀,王新强,高婷婷,等.Cu、Ag和Au掺杂AlN的电磁性质的第一性原理研究[J].原子分子物理学报,2012,29(1):0167-0175.
[32]Wu R Q,Shen L,Yang M,et al.Enhancing hole concentration in AlN by Mg:O codoping:Ab initio study[J].Phy Rev B,2008,77(7):73203-73207.
[33]Suzuki M,Uenoyama T,Yanase A.First-principles calculations of effective-mass parameters of AlN and GaN[J].Phys Rev B,1995,52(11):8132.
[34]Peng H W,Xiang H J,Wei S H,et al.Origin and enhancement of hole-induced ferromagnetism in first-row d0semiconductors[J].Phys Rev Lett,2009,102(1):017201-017204.
[35]沈学础.半导体光谱和光学性质[M].第二版.北京:科学出版社.2003:P76.
[36]邢海英,范广涵,章勇,等.第一性原理研究Mg,Si和Mn共掺GaN[J].物理学报,2009,58(1):450-458.
[37]侯清玉,张越,张涛.含氧空位锐钛矿TiO2光学性质的第一性原理研究[J].光学学报,2008,28(7):1347-1353.
First-principles Study of Optical and Electronic Properties of Ag Doped AlN Semiconductors
DENG Junquan,WU Zhimin,WANG Ailing,ZHAO Ruoyu,HU Aiyuan
(College of Physics and Electronic Engineering,Chongqing Normal University,Chongqing 401331,China)
Geometrical structure of Ag doped 32-atom super-cell of AlN was optimized with first principle density functional theory based on full potential linearized augumented plane wave method.Electronic structures,magnetic and optical properties were calculated and discussed in detail.It shows that Ag doping makes Ag4d electrons hybrid with its nearest neighbor N2p electrons,which introduces impurity bands to form acceptor energy level and realizes p-type doping.The system enhances its conductivity and shows metallic and weak magnetism.Its net magnetic moment is 1.38 μв.Mulliken charge population of N-Ag bonds through Ag doping is small and bonds show strong ionic bonding properties.Imaginary part of dielectric function and optical absorption spectrum of the doping system show a new peak in low energy region.Complex refractive index function changes in low energy region.And absorption edge extends to low energy.Doping system enhances long-wave absorption and energy loss decreases obviously.
Ag-doped AlN;electronic structures;ferromagnetism;optical properties;first-principles
date:2013-10-12;Revised date:2014-01-26
O469;O472
A
2013-10-12;
2014-01-26
国家自然科学基金(61201119)、教育部科学技术重点项目(211152)、重庆市教委科研项目(KJ110634)和国家创新创业训练计划(201310637001)资助项目
邓军权(1989-),男,本科生,主要从事半导体功能材料的研究,E-mail:dengjq2014@163.com
*通讯作者:毋志民,E-mail:zmwu@cqnu.edu.cn
1001-246X(2014)05-0617-08
