助剂对Mo2C/MoO2-Zr O2催化剂异构化性能的影响
2014-06-09王海彦宋丽娟孙兆林
施 岩, 王海彦, 宋丽娟, 孙兆林
(1.中国石油大学(华东),山东东营266580;2.辽宁石油化工大学化学化工与环境学部,辽宁抚顺113001)
助剂对Mo2C/MoO2-Zr O2催化剂异构化性能的影响
施 岩1,2, 王海彦1,2, 宋丽娟1,2, 孙兆林1,2
(1.中国石油大学(华东),山东东营266580;2.辽宁石油化工大学化学化工与环境学部,辽宁抚顺113001)
研究了Mo2C/Mo O2-Zr O2催化剂上正己烷的异构化反应。分别以十六烷基三甲基溴化铵和聚乙二醇20000为模板剂,采用共沉淀法制备前驱体固体超强酸Mo O3-Zr O2,从前驱体MoO3-Zr O2的BET和FTIR表征可以看出,所得样品具有适宜比表面积、孔径和孔容。前驱体MoO3-Zr O2经碳化后总酸量大幅度的下降。加入混合助剂的Mo2C/MZ-P0.005-C0.5的催化剂无论转化率和选择性都要比加入单一助剂的催化剂要高得多,其转化率达67.5%,选择性75.2%。
异构化; 模板剂; 前驱体
近年来研究发现,过渡金属的磷化物催化剂具有较高的加氢脱氮活性和稳定性,且催化剂具有较强的抗硫中毒性能,催化剂的加氢选择性远高于传统的商业用催化剂,过渡金属磷化物催化剂的氢耗低、氢能利用率较高。过渡金属磷化物中Ni2P的加氢脱氮活性和加氢脱硫的催化性能最佳,远高于其他过渡金属磷化物,非负载的Ni2P催化剂比表面积相对较小,还不足1 m2/g,需将其负载在具有较大比表面积的载体上,以提高催化剂的活性表面积,过渡金属磷化物作为一种新型的催化材料,无论是加氢脱硫还是加氢脱氮都有比硫化物催化剂更加优越的潜力[1-2]。
碳化钼与贵金属有相似的表面性质和催化特性引起了人们的广泛关注,碳化钼已经广泛应用于催化加氢、催化脱氢、催化加氢脱硫、催化加氢脱氮、异构化和芳构化等反应过程[3]。本实验以十六烷基三甲基溴化铵(CTAB)和聚乙二醇(PEG)20000为模板剂,采用共沉淀法制备前驱体固体超强酸前驱体MoO3-Zr O2,然后负载Mo2C制备了Mo2C/MoO2-Zr O2催化剂并应用于正己烷的异构化反应。从MoO3-Zr O2的BET和FTIR表征可以看出,所得样品具有适宜比表面积、孔径和孔容,前驱体Mo O3-Zr O2经碳化后总酸量大幅度下降。
1 实验部分
1.1 实验试剂
正己烷,氧氯化锆、钼酸铵、硝酸钯、氢氧化氨、聚乙二醇20000、十六烷基三甲基溴化铵(分析纯,国药集团化学试剂有限公司)。
1.2 催化剂的制备
前驱体固体超强酸Mo O3-Zr O2的碳化在自制的连续式小型固定床反应装置上进行,在不锈钢反应管中部装入固体超强酸Mo O3-Zr O2。首先在H2压力为0.5 MPa、温度为500℃、H2流速10 m L/s的条件下还原2 h,Mo O3-Zr O2转变为Mo O2-Zr O2,然后以正己烷为碳源,采用程序升温还原法进行碳化,100~300℃升温速率2℃/min,300~650℃升温速率1℃/min,并在650℃下保持1 h。正己烷的进料速率使用双柱塞微量计量泵控制在0.33 m L/min,H2流速10 m L/s。最后在H2压力0.5 MPa、H2流速10 m L/s、温度600℃下临氢还原1 h,即制得Mo2C/Mo O2-Zr O2催化剂[4]。
通过等体积浸渍法将一定量氯铂酸负载到固体超强酸Mo O3-Zr O2上[5]。静置12 h后,110℃干燥4 h,以2℃/min的升温速率升至550℃,恒温焙烧4 h,即制得质量分数0.5%的Pt/Mo O3-Zr O2催化剂,用于与Mo2C/MoO2-Zr O2催化剂进行比较。
1.3 催化剂的表征
催化剂样品的晶相结构使用日本岛津Shimadzu XRD-6000型转靶X射线衍射仪上测定;样品FTIR表征采用Nicolet Impact 410型红外光谱仪进行红外分析;样品的N2吸附脱附等温线在美国Micromeritics Instrument Corporation生产的ASAP2405型自动物理吸附仪上进行测试,通过低温(-196℃)N2吸附,利用BET法测定样品的比表面积,静态容量法测定孔容和BJH法计算孔径分布。FTIR测定选用Perkin-Elmer公司生产的Spectrum TM GX傅里叶变换红外光谱仪,中红外DTG检测器,KBr制样。
2 结果与讨论
2.1 助剂对前躯体和催化剂孔结构的影响
助剂对前躯体和催化剂的比表面积和孔结构影响较大,对应的助剂配比不同,制备了一系列固体超强酸Mo O3-Zr O2,记为MZ-Px-Cy(其中x代表PEG与锆原子的物质的量比,y代表CTAB与锆原子的物质的量比,例如MZ-P0.005-C0.5为PEG和 CTAB与锆离子的物质的量比分别为0.005和0.5),其Mo的负载量都为20%。
在制胶过程中,CATB在水中会形成球形胶束,在形成的胶束中亲水基形成了外表面,僧水基朝着中心。这样利用存在着的空间位阻效应和静电效应减少了尾部与水的副反应,但是却引入了带电荷的端基的相互排斥竞争的副反应。这个相互竞争的副反应的平衡能使胶体处于均匀分散状态,控制团聚提高比表面积。助剂的浓度决定了胶束的形状、胶束之间的稳定性和胶束化的程度;有机共聚物的助剂可以通过改变其本身的化学结构、链长、官能团,达到调节样品的孔结构、机械性能和热稳定性能的目的,从而合成出理想的介孔分子筛。加入适量的PEG溶液,其烷氧基能取代Zr4(OH)b8(OH)t8(H2O)x水凝胶胶粒表面的非架桥羟基(OH)t,消除氢键作用引起的硬团聚,起到一定的分散作用。
表1为前躯体和催化剂的织构参数。由表1可以看出,当只加入助剂CTAB时,样品MZ-P0-C0.5比表面积最大,孔径、孔容最小。当只加入助剂PEG时,样品MZ-P0.005-C0比表面积减少,孔径、孔容增大。当加入助剂CTAB和PEG的混合溶液时,样品MZ-P0.005-C0.5比表面继续减小,而孔径、孔容增大,这可能是由于模板剂溶液浓度过大,以至样品的孔壁过薄,高温焙烧时塌陷导致比表面积减少。而以样品MZ-P0.005-C0.5为前躯体经程序升温碳化后制成的催化剂Mo2C/MZ-P0.005-C0.5,其比表面积最小,孔径最大。这可能是由于碳化过程中的积碳填充前躯体MZ-P0.005-C0.5表面的沟壑,并有可能堵塞了小孔,造成了比表面积由71.60 m2/g减少到42.59 m2/g,几乎减少了一半。

表1 助剂对前躯体和催化剂的比表面积、孔径和孔体积的影响Table 1 Effect of the surfactants on surface area,pore size and pore volume of precursor and catalyst samples
前躯体的比表面积和孔体积的变化与其孔结构相关。图1为制备的前躯体N2吸附脱附曲线。图1(a)为样品MZ-P0-C0.5的N2吸附脱附曲线。由图1(a)可知,吸附脱附曲线为典型的Ⅳ类曲线,其在p/p0=0.45~0.8出现突越,具有H2型迟滞环。这表明样品MZ-P0-C0.5具有典型的介孔特征,其孔型主要为墨水瓶孔道结构。图1(b)为样品MZ-P0.005-C0的N2吸附脱附曲线,其吸附脱附等温线,与样品MZ-P0-C0.5相似,同为Ⅳ类曲线,其p/p0=0.4~0.9出现H2型迟滞环,其在高压区域基本没有表现出吸附限制,说明有一定的大孔形成。图1(c)为样品MZ-P0.005-C0.5的N2吸附脱附曲线,在低分压阶段(p/p0<0.4),N2的吸附量随分压的升高近似线性增加。这是由于N2在孔壁上发生单层或多层吸附引起的[6]。随着压力增加,当p/p0>0.4时,曲线上翘,吸附量突增,当p/p0=0.45~0.9出现H3型迟滞环,其原因为N2在骨架围成的介孔内产生的毛细管凝聚的结果,且其在高压区域并未表现出吸附限制。这表明其孔径分布较广,其孔型多为裂口型孔道[7]。
图1(d)为以样品MZ-P0.005-C0.5为前躯体碳化后的催化剂的N2吸附脱附曲线。由图1(d)可以看出,其吸附脱附等温线为Ⅳ类曲线,p/p0=0.75~1.0出现H3型迟滞环,且其p/p0<0.4的等温线斜率非常小,这说明此样品几乎没有微孔结构[8]。这进一步说明了,碳化过程中生成的积碳堵塞了微孔的假设。
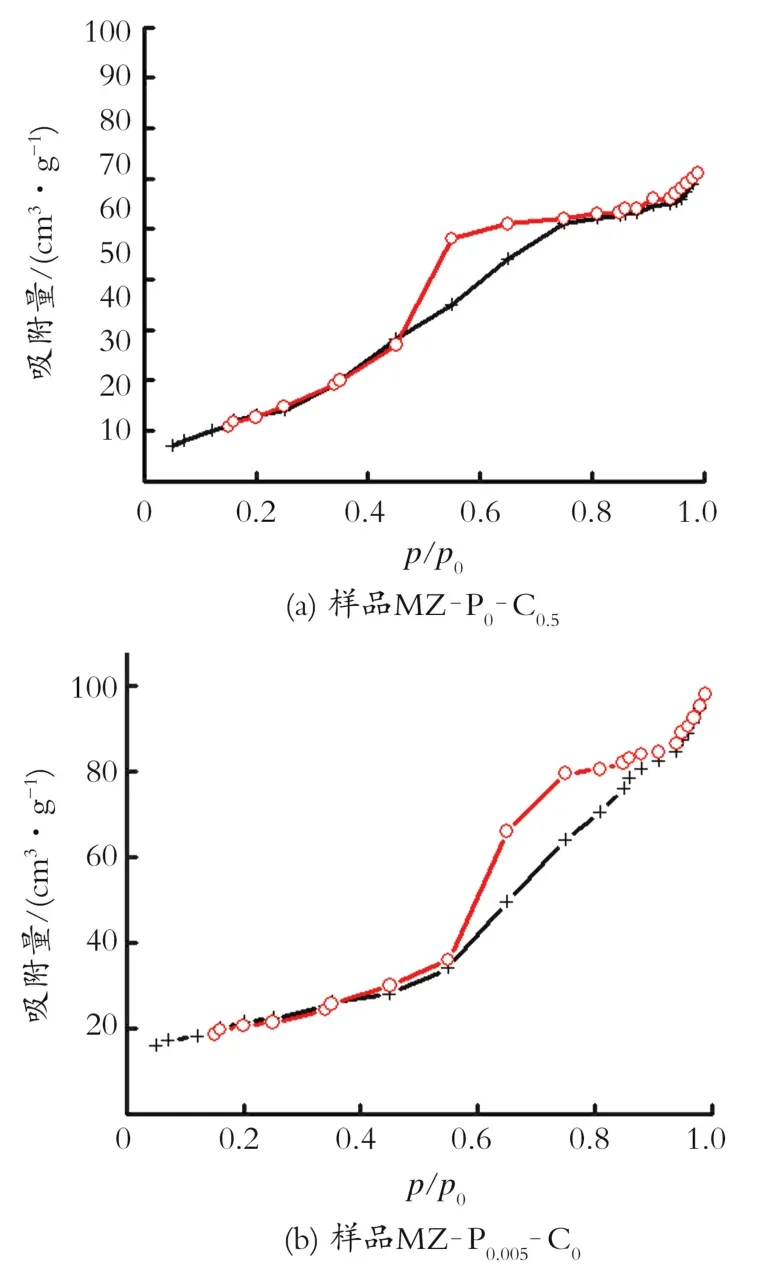

图1 制备的前驱体和催化剂样品的BET比表面积和N2吸附脱附曲线Fig.1 The BET N2adsorption and desorption curve of precursor and catalyst sample
图2为前躯体和催化剂样品的孔径分布。由图2可以看出,只加入CTAB一种助剂的样品MZ-P0-C0.5的孔径在2~8 nm,只加入PEG的样品MZP0.005-C0的孔径在3~10 nm,当加入CTAB和PEG混合助剂后样品MZ-P0.005-C0.5的孔径主要分布在2~10 nm,但却有20~45 nm的较大孔出现,而碳化后的催化剂样品Mo2C/MZ-P0.005-C0.5的孔径增大至4.8~25 nm,同时孔径在25~45 nm也有分布。
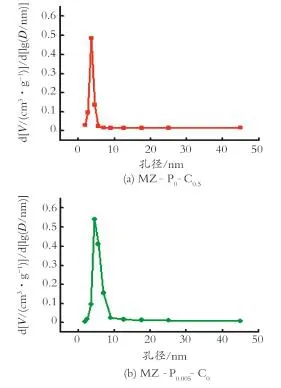

图2 前躯体和催化剂样品的孔径分布Fig.2 Pore size distribution of precursor and catalyst samples
2.2 助剂对催化剂及正己烷异构化活性的影响
在反应温度380℃、反应压力2.5 MPa、氢烃体积比400∶1、体积空速1 h-1的条件下考察助剂对Mo2C/MZ系列催化剂的异构化反应性能的影响,结果见表2。

表2 助剂对Mo2C/MZ催化剂异构化活性的影响Table 2 Effect of the surfactants on n-hexane isomerization over the Mo2C/MZ catalysts %
从表2可以看出,单一加入助剂CATB的Mo2C/MZ-P0-C0.5催化剂和单一加入助剂PEG的Mo2C/MZ-P0.005-C0催化剂相比,Mo2C/MZ-P0-C0.5催化剂的转化率要高,但选择性要低10%左右,而加入混合助剂的Mo2C/MZ-P0.005-C0.5的催化剂无论转化率和选择性都要比加入单一助剂的催化剂要高得多,其转化率达67.5%,选择性达75.2%。
在反应温度380℃、反应压力2.5 MPa、氢烃体积比400∶1、体积空速1 h-1的条件下助剂对Pt/ MZ系列催化剂的异构化反应性能的影响见表3。
由表3可知,助剂对Pt/MZ系列催化剂的异构化活性影响与Mo2C/MZ系列催化剂相似。Pt/MZ系列催化剂同样是加入混合助剂的催化剂异构化活性最佳,转化率达70.1%,选择性达82.5%。加入单一的CTAB的催化剂比表面积最大,孔径最小;其次是加入单一的PEG的催化剂;比表面积最小的是加入混合助剂的催化剂,但其孔径最大。从异构化活性上看,恰恰是比表面积最小、孔径最大的Pt/ MZ-P0.005-C0.5催化剂异构化活性最佳。虽然大的比表面积可能是活性组份分散的更均匀且可以增大原料与活性中心接触的几率[9]。但由数据可知,比表面积并不是唯一的影响因素。其中孔结构对催化剂的影响也至关重要。随着孔径的增大异构化选择性也逐渐提高,这可能是由于当正己烷在催化剂的活性中心上进行异构化反应,生成单支链或多支链的异构体,其分子直径较大,可能导致其无法通过催化剂孔道,这加剧了二次反应导致裂解率高,选择性下降。所以孔径较小的墨口瓶型孔道的催化剂Pt/ MZ-P0-C0.5和Pt/MZ-P0.005-C0要比裂口型孔道的催化剂Pt/MZ-P0.005-C0.5活性要弱很多。

表3 助剂对Pt/MZ催化剂异构化活性的影响Table 3 Effect of the surfactants on n-hexane isomerization over the Pt/MZ catalysts %
综上,确定采用助剂CTAB和PEG的混合溶液(nPEG/nZr=0.005,nCTAB/nZr=0.5)来制备前躯体MoO3-Zr O2。
3 结论
以十六烷基三甲基溴化铵和聚乙二醇20000为模板剂,采用共沉淀法合成前驱体固体超强酸Mo O3-Zr O2,负载活性组分碳化钼制备出Mo2C/ Mo O2-Zr O2催化剂,并以正己烷为原料进行异构化反应。从前驱体Mo O3-Zr O2的表征结果可看出,所得样品具有适宜比表面积、孔径和孔容。前驱体Mo O3-Zr O2经碳化后总酸量大幅度的下降。最终确定将助剂CTAB和PEG的混合溶液(nPEG/nZr= 0.005,nCTAB/nZr=0.5)作为模板剂来制备前躯体Mo O3-Zr O2效果最佳。
[1] 李学斌,常勇,王海彦,等.正庚烷在β沸石负载碳化钼催化剂上的异构化研究[J].燃料化学学报,2007,35(2):228-332.
Li Xuebin,Chang Yong,Wang Haiyan,et al.n-Heptane isomerization in beta zeolite[J].Journal of Fuel Chemistry and Technology,2007,35(2):228-332.
[2] 李旭,王昕,施力,等.正己烷异构化Pd/Beta催化剂的研究[J].石油炼制与化工,2004,35(7):16-20.
Li Xu,Wang Xin,Shi Li,et al.Study on n-hexane isomerization catalyst Pd/Beta[J].Petroleum Processing and Chemical Industry,2004,35(7):16-20.
[3] Boskovic G,Micic R,Pavlovic P,et al.n-Hexane isomerization over Pt-Na(H)Y catalysts obtained by different preparation methods[J].Catalysis Today,2001,65(2-4):123-128.
[4] Tiitta M,Harlin E,Makkonen J,et al.Medium and large pore zeolites in n-hexene skeletal isomerization[J].Studies in Surface Science and Catalysis,2004,154(3):2323-2330.
[5] Villegas J I,Kumar N,HeikkiläT,et al.A highly stable and selective Pt-modified mordenite catalyst for the skeletal isomerization of n-butane[J].Applied Catalysis A:General,2005,284(1-2):223-230.
[6] 蔡国辉,肖益鸿,詹瑛瑛,等.表面分散剂对Pt/Zr O2催化剂结构的影响[J].福州大学学报:自然科学版,2003,31(2): 238-242.
Cai Guohui,Xiao Yihong,Zhan Yingying,et al.Effect of dispersant on the surface structure of Pt/Zr O2catalysts[J]. Journal of Fuzhou University:Natural Science Edition,2003,31(2):238-242.
[7] 汪颖军,张海菊,田性刚,等.Co/WO3/Zr O2催化正辛烷临氢异构化反应性能[J].石油学报,2009,25(4):490-495.
Wang Yingjun,Zhang Haiju,Tian Xinggang.Co/WO3/Zr O2catalytic n-octane hydroisomerization performance of[J]. Acta.Petrolei.Sinica.,2009,25(4):490-495.
[8] Belskaya O B,Danilova I G,Kazakov M O,et a1.Investigation of active metal species formation in Pd-promoted sulfated zirconia isomerization catalyst[J].Applied Catalysis A:General,2010,387(1-2):5-12.
[9] 徐占林,王良,赵丽娜,等.固体超强酸S/Zr O2-Al2O3催化正丁烷异构化反应研究[J].石油与天然气化工,2006,35 (1):13-16.
Xu Zhanlin,Wang Liang,Zhao Lina,et al.S/Zr O2-Al2O3solid superacid catalyst n-butane isomerization reaction of [J].Chemical Engineering of Oil and Gas,2006,35(1):13-16.
(编辑 闫玉玲)
Effect of Additives on the Isomerization Performance of Mo2C/MoO2-Zr O2Catalyst
Shi Yan1,2,Wang Haiyan1,2,Song Lijuan1,2,Sun Zhaolin1,2
(1.China University of Petroleum(Huadong),Dongying Shandong 266580,China; 2.College of Chemistry,Chemical Engineering and Environmental Engineering,Liaoning Shihua University,Fushun Liaoning 113001,China)
The isomerization of n-hexane over Mo2C/MoO2-Zr O2catalyst was investigated.First,the precursor solid superacid MoO3-Zr O2was prepared by coprecipitation process,using the mixture of cetyltrimethylammonium bromide and polyethylene glycol 20000 as templates.It can be seen that the precursor samples had applicable surface area,pore diameter and volume from the BET and FTIR characterization of the Mo2C/MoO2-Zr O2.The total acid of precursor MoO3-Zr O2had a large decline after carbonized.The conversion and selectivity of Mo2C/MZ-P0.005-C0.5are much more than single auxiliary catalystafter adding catalyst mixed assistant.The conversion rate and selectivity reached 67.5%,75.2%,respectively.
Isomerization;Template;Precursor
TE624.9+4;O643.32
A
10.3969/j.issn.1006-396X.2014.03.009
1006-396X(2014)03-0036-05
2013-08-26
2014-03-16
国家自然科学基金(20976077,21076100)。
施岩(1977-),男,博士研究生,讲师,从事清洁燃料生产工艺研究;E-mail:shiyan1816@163.com。
