乙烷热裂解自由基反应机理的综合数值模拟*
2014-06-09张红梅李金莲郝玉兰
张红梅,姜 维,李金莲,郝玉兰,赵 亮
(1.东北石油大学 石油与天然气化工省重点实验室,黑龙江 大庆 163318;2.中国石油大学 重质油国家重点实验室,北京 102249)
烃类蒸汽热裂解是生产3大基础烯烃乙烯、丙烯和丁二烯的重要手段[1]。烯烃生产行业的剧烈竞争使得原料及生产过程的优化显得极其重要[2-4]。计算机模拟技术以其计算速度快、适应原料频繁变动的优点成为热裂解过程优化和设计越来越主要的手段[5]。热裂解过程的化学反应属于自由基反应机理,因此得到不同原料准确的自由基反应规律是优化计算的前提。
作者在对现有自由基机理研究方法的局限性进行分析的基础上,提出了将分子模拟[6-9]和管式反应器工艺模拟[10]相结合,来了解单一原料烃裂解反应机理的新方法,并以乙烷热裂解为例对该方法进行了介绍,同时得到了乙烷热裂解反应产物及自由基的详细反应规律。
1 对现有自由基机理研究方法的剖析
一般而言,对某一烃类分子进行自由基机理研究采用如下的方法:首先对该烃类分子进行实验,通过对实验结果的分析及总结,得到该分子进行热裂解可能发生的自由基反应,并通过实验测定或计算这些自由基反应的活化能和指前因子[11];其次以上述实验数据为动力学模型,假设在反应达到稳态时各自由基产生和消失的速度相等,得到不同自由基的非线性微分方程组;最后用微分方程的数值解法Treanor法[12-13]、Gear法[13-14]或拟稳态法[15-17]等对非线性方程组进行求解,可得到各自由基沿管长的变化规律,进而可得到各产物的分布。
作者曾用上述方法对乙烷热裂解反应机理进行了研究[18]。首先用分子模拟软件Materials Studio[19-20]对乙烷热裂解可能发生的主要反应进行分子模拟计算,得到的计算结果见表1。

表1 乙烷热裂解各基元反应及动力学数据
将上述自由基进行组合,可设计得到不同的反应路径,下面是经配平后的一个路径。
反应路径1,由表1中的反应(2)、(3)、(4)、(5)和(7)构成。

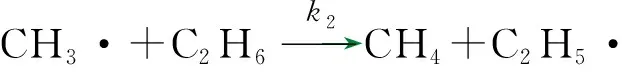
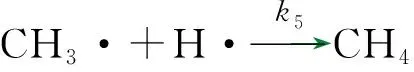
对于自由基链式反应,由于自由基活性很大,所以存在的浓度很小,可以假定在稳态时自由基浓度不随时间而变,即任意一个自由基的生成速度等于其消失速度[21],由此可以推导出各自由基在稳态条件下的物料平衡方程式。以下分别是甲基、氢和乙基自由基的物料平衡方程式。

将[H·]、[CH3·]和[C2H5·]看做未知数,解上述方程组,得:

由此可得产物乙烯的生成速率为:
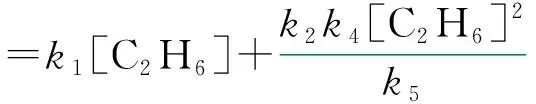
由上述推导可以看出,如将活化能大的反应舍去,第一部分的活化能偏小,与实验值不符。同样可设计得到路径2~4。
反应路径2由表1中的反应(2)、(3)、(4)、(5)和(6)构成;反应路径3由表1中的反应(2)、(3)、(4)、(5)和(7)构成;反应路径4由表1中的反应(2)、(4)、(5)和(7)构成。采用与反应路径1同样的方法,可得到路径1~4的活化能见表2。
根据文献[22]中的实验数据可知,实际测得乙烷热裂解的反应活化能为263.6~293.7 kJ/mol。上述结果都与实验结果不符。
对上述结果进行分析可以看出,经典数值方法机理模型的数学表达式为一常微分方程组,求解该常微分方程组是这一复杂的机理模型能否成功的关键。而由上述推导方法可以看出,该方法存在着配平的任意性、主观性,且不同的配平方法将得到不同的结果,这是经典数值方法的局限性。
2 乙烷热裂解机理的数值模拟计算方法
采用分子模拟得到的乙烷热裂解自由基机理反应动力学模型代入自建的管式炉一维模型中进行工艺模拟计算的方法,对乙烷热裂解反应规律进行研究,通过自由基和产物沿管长的变化数据探讨研究乙烷热裂解的详细反应机理。
将乙烷热裂解不同自由基反应的分子模拟计算结果进行分析整理,可得到如图1所示的2条反应路径。
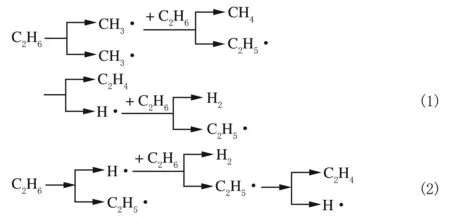
图1 乙烷热裂解自由基反应路径
其中路径1是乙烷C—C键断裂所引发的反应,路径2是C—H键断裂所引发的反应。
研究内容包括3个方面:(1)将上述所有自由基反应作为动力学模型进行工艺模拟计算(简称总路径),观察各自由基浓度在反应器内沿管长的变化情况;(2)在同样操作条件下分别对路径1和路径2进行模拟计算;(3)对计算结果进行分析。
将所有自由基反应作为动力学模型代入工艺模型中进行模拟计算,各自由基和各产物摩尔分数沿管长的变化规律见图2和图3。
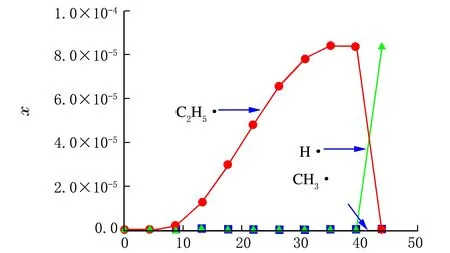
管式裂解炉长度/m
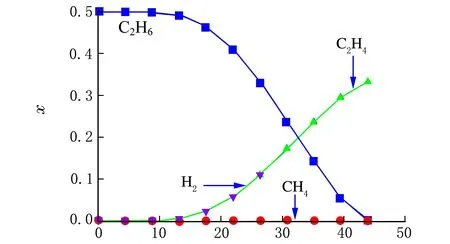
管式裂解炉长度/m
对路径1进行计算,可得到与图2和图3相同的结果;对路径2进行计算,计算结果见图4所示。由图4可以看出,同样温度条件下,路径2中乙烷几乎不发生裂解反应。
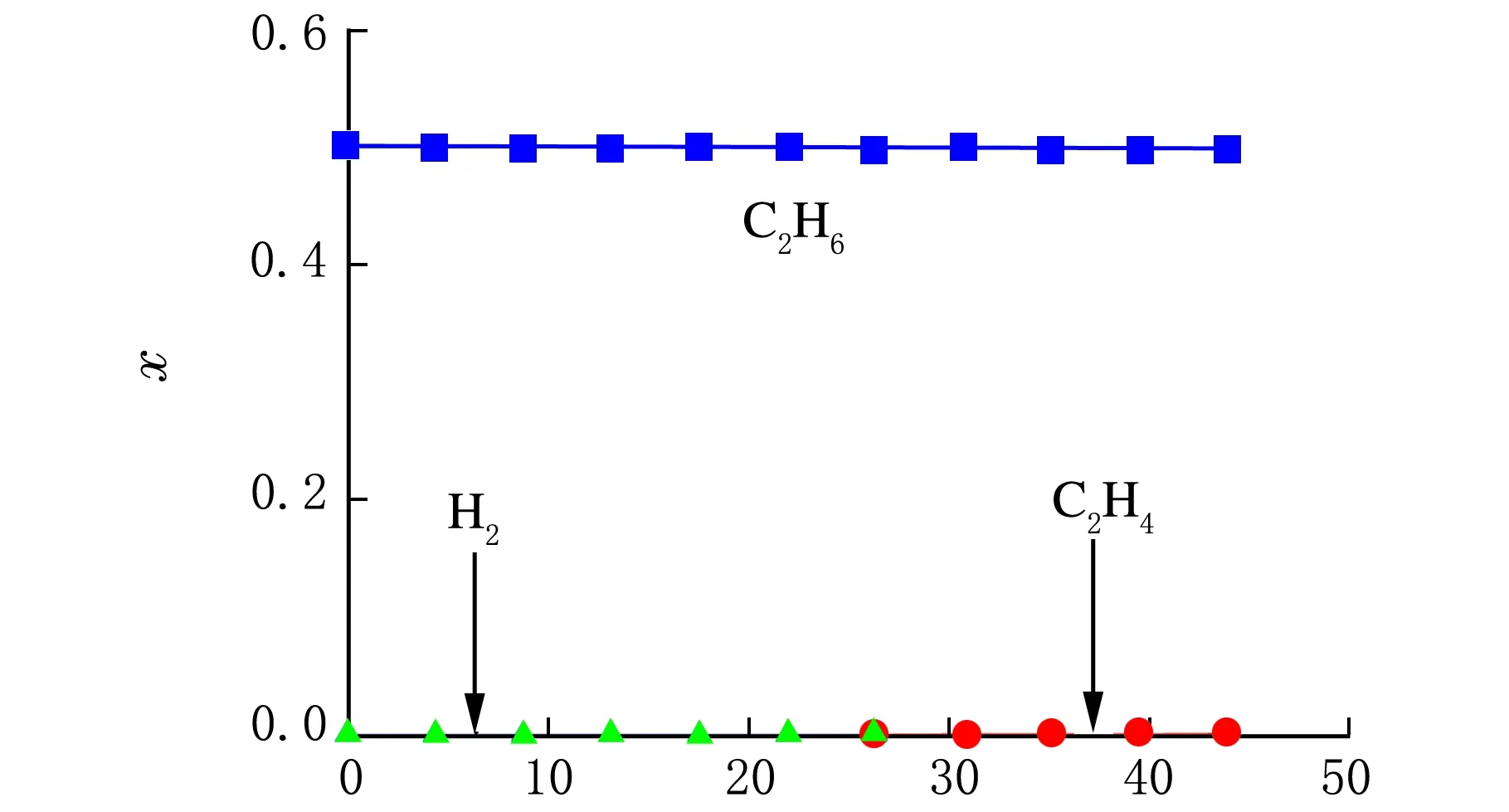
管式裂解炉长度/m
由上述计算可知,乙烷热裂解反应以断C—C键为主,C—H键断裂几乎不发生反应。
3 乙烷热裂解模拟计算结果分析
对比图2和图3中的各自由基和产物沿管长的变化规律进行分析,可得到乙烷热裂解的详细反应机理,现说明如下。
在反应开始阶段,反应温度较低,未达到C—C键、C—H键的断键温度,因此C2H6维持进料浓度不变;随着温度的上升,由于此时没有自由基生成,且乙烷分子中C—C键断裂所需的活化能为417 kJ/mol,比C—H键断裂所需的活化能451 kJ/mol小,故C—C键断裂比C—H键断裂要容易得多,因此反应管外传递的热量只能提供给乙烷分子断C—C键,生成少量的甲基自由基CH3·。
少量的活泼CH3·迅速与乙烷进行链转移反应生成甲烷和乙基自由基而被消耗掉,管内又处于没有自由基的状态,因此能量只能提供给乙基自由基进行分解反应,生成乙烯和氢自由基;活泼的H·迅速与乙烷反应生成氢气和C2H5·,C2H5·继续分解生成C2H4和H·,形成了一个可持续反应的链传递循环过程;在此阶段,乙烷被不断地消耗,乙烯和氢气不断生成。而甲烷只有链引发时有极少量的生成。
随着链传递反应的不断循环进行,乙烷不断减少,导致链传递所需的氢自由基减少,因此氢自由基与乙烷的链传递受乙烷减少的影响而减少,因此能量只能提供给C2H5·分解反应,这就是在管长约39.5米以后,C2H5·迅速减少,而H·大量生成的原因。
由上述分析可知,乙烷链终止反应主要由2个氢自由基生成氢气为主。
4 结 论
(1) 利用实验方法研究烃类热裂解反应存在着人力、物力消耗大,实验结果影响因素复杂、难以得到具体自由基反应机理的缺点;传统的“假设在反应达到稳态时各自由基产生和消失的速度相等”研究烃类热裂解自由基反应机理的方法存在较多的任意性和人为因素,造成了计算结果的局限性,使得计算结果与实验结果不符,影响了对烃类热裂解机理的研究。
(2) 作者提出了用分子模拟软件研究烃类热裂解的自由基基元反应,将其计算结果作为动力学模型,代入自建的一维工艺数学模型进行模拟计算,通过对计算得到的自由基和产物沿管长的变化规律的分析,提出了了解烃类热裂解反应机理的新方法,并通过对乙烷热裂解机理的研究证明了该方法的可行性。
(3) 乙烷热裂解反应机理如下:链引发反应主要由断C—C键生成2个甲基自由基完成,但由于后续链传递反应没有甲基自由基生成,因此甲基自由基没有参与到链传递反应中,生成的甲烷数量很少;乙烷的链传递反应主要由氢自由基与乙烷生成乙基自由基和氢气、乙基自由基分解生成乙烯和氢自由基为主反复循环进行;链终止反应主要由两个氢自由基生成氢气为主。
[ 参 考 文 献 ]
[1] 杜志国,王国清,李蔚,等.烃类蒸汽裂解反应动力学模型进展[J].乙烯工业,2010,22(1):12-16.
[2] 张红梅,张晗伟,顾萍萍,等.丙烷热裂解反应机理的分子模拟[J].石油学报(石油加工),2012,28(6):146-150.
[3] 何小龙.茂名石化公司乙烯原料的优化[J].石油化工,2009,38(1):64-67.
[4] 万书宝,贺德福.蒸汽裂解制乙烯的发展趋势[J].现代化工,2009,29(6):6-10.
[5] 李金莲,张红梅,张晗伟,等.提高蒸汽热裂解三烯收率最佳操作条件的模拟计算[J].石油学报(石油加工),2013,29(4):694-699.
[6] Aribike D S,Susu A A.Mechanistic modeling of the pyrolysis ofn-heptane[J].Thermochimica Acta,1988,127(16):259-273.
[7] LU L H,LU X H,CHEN Y P,et al.Monte carlo simulation of adsorption of binary and quaternary alkane isomers mixtures in zeolites:effect of pore size and structure[J].Fluid Phase Equilibria,2007,259(2):135-145.
[8] Ungerer P,Nieto-Draghi C,Rousseau B,et al.Molecular simulation of the thermophysical properties of fluids:from understanding toward quantitative predictions[J].Journal of Molecular Liquids,2007,134:71-89.
[9] 胡瑶,杨晓宁.ScCO2溶剂中金纳米颗粒界面性质的分子模拟[J].化工学报,2011,62(2):295-300.
[10] 张红梅,贺永殿,蓝兴英,等.管式裂解炉管二维工艺模型研究新进展及应用[J].化工进展,2006,25(7):791-794.
[11] 李蔚,张兆斌,周丛,等.裂解炉管内自由基反应模型的研究进展[J].乙烯工业,2010,22(2):1-6.
[12] 魏巍.SRT-Ⅳ(HC)型裂解炉中HVGO裂解过程的模型化与模拟[D].北京:北京化工大学,2006:38-40.
[13] 徐士良.C常用算法程序集[M].北京:清华大学出版社,1994:200-213.
[14] 倪力军,张立国,倪进方,等.乙烷工业炉裂解动力学模型与模拟[J].石油学报(石油加工),1995,13(4):70-77.
[15] Joo Eunjung,Park Sunwon.Pyrolysis reaction mechanism for industrial naphtha cracking furnaces[J].Ind Eng Chem Res,2001,40:2409-2415.
[16] Blakemore J E,Coreonan W H.Validity of the steady-state approximation applied to the pyrolysis ofn-butane[J].I&EC Proc Des Dev,1969,8(2):206-209.
[17] Tomlin Alison S,Pilling M J H,et al.Reduced mechanisms for propane pyrolysis[J].Ind Eng Chem Res,1995,34(11):3749-3760.
[18] 郝玉兰,张红梅,张晗伟,等.丁烷热裂解反应机理的分子模拟[J].石油学报(石油加工),2013,29(5):68-73.
[19] 吴健.Materials studio在结构化学教学中的一些应用[J].高校实验室工作研究,2007,3:47-48.
[20] 庄昌清,岳红,张慧军.分子模拟方法及模拟软件Materials Studio在高分子材料中的应用[J].塑料,2007,39(4):81-84.
[21] 邹仁鋆.石油化工裂解原理与技术[M].北京:化学工业出版社,1982:76-80.
[22] 吴指南.基本有机化工工艺学[M].北京:化学工业出版社,1990,23-30.
