Overexpression of C-terminal fragment of glutamate receptor 6 prevents neuronal injury in kainate-induced seizure via disassembly of GluR6-PSD95-MLK3 signaling module
2014-06-01JieMouXiaomeiLiuDongshengPei
Jie Mou, Xiaomei Liu, Dongsheng Pei
1 Jiangsu Key Laboratory of Targeted Drug and Clinical Application, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
2 School of Basic Medical Science, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
3 Jiangsu Key Laboratory of Biological Cancer Therapy, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
Overexpression of C-terminal fragment of glutamate receptor 6 prevents neuronal injury in kainate-induced seizure via disassembly of GluR6-PSD95-MLK3 signaling module
Jie Mou1, Xiaomei Liu2, Dongsheng Pei3
1 Jiangsu Key Laboratory of Targeted Drug and Clinical Application, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
2 School of Basic Medical Science, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
3 Jiangsu Key Laboratory of Biological Cancer Therapy, Xuzhou Medical College, Xuzhou, Jiangsu Province, China
Jie Mou and Xiaomei Liu contributed equally to this study.
Our previous study showed that when glutamate receptor (GluR)6 C terminus-containing peptide conjugated with the human immunodeficiency virus Tat protein (GluR6)-9c is delivered into hippocampal neurons in a brain ischemic model, the activation of mixed lineage kinase 3 (MLK3) and c-Jun NH2-terminal kinase (JNK) is inhibited via GluR6-postsynaptic density protein 95 (PSD95). In the present study, we investigated whether the recombinant adenovirus (Ad) carrying GluR6c could suppress the assembly of the GluR6-PSD95-MLK3 signaling module and decrease neuronal cell death induced by kainate in hippocampal CA1 subregion. A seizure model in Sprague-Dawley rats was induced by intraperitoneal injections of kainate. The effect of Ad-Glur6-9c on the phosphorylation of JNK, MLK3 and mitogen-activated kinase kinase 7 (MKK7) was observed with western immunoblots and immunohistochemistry. Our findings revealed that overexpression of GluR6c inhibited the interaction of GluR6 with PSD95 and prevented the kainate-induced activation of JNK, MLK3 and MKK7. Furthermore, kainate-mediated neuronal cell death was signi fi cantly suppressed by GluR6c. Taken together, GluR6 may play a pivotal role in neuronal cell death.
nerve regeneration; brain injury; hippocampal neuronal injury; seizures; adenovirus; GluR6; PSD95; MLK3; kainate; apoptosis; JNK; NSFC grants; neural regeneration
Funding: This work was supported by the National Natural Science Foundation of China, No. 30800309, 81372172; the Educational Science Foundation of Jiangsu Province, China, No. 10KJB350005; the Xuzhou Science Foundation in China, No. XZZD1153; the President Special Grant of Xuzhou Medical College in China, No. 09KJZ20; and a grant from the Zhenxing Project Foundation of XZMC.
Mou J, Liu XM, Pei DS. Overexpression of C-terminal fragment of glutamate receptor 6 prevents neuronal injury in kainate-induced seizure via disassembly of GluR6-PSD95-MLK3 signaling module . Neural Regen Res. 2014;9(23):2059-2065.
Introduction
Kainate receptors mediate the majority of excitatory synapse transmissions in the mammalian central nervous system, thereby exerting key effects on synaptic plasticity as well as in pathological processes such as ischemia and epilepsy (Dingledine et al., 1999). Kainate receptors also have varied patterns of expression in the subregions of the hippocampus (Bureau et al., 1999). GluR6 is mainly located in the CA1 and CA3 regions, and plays a signi fi cant role in learning and memory (Darstein et al., 2003). GluR6-de fi cient rodents are resistant to kainate-induced excitotoxicity, suggesting that GluR6 likely mediates the neurotoxic effect of glutamate (Mulle et al., 1998). Studies in cerebral ischemia have confi rmed that a correlation exists between GluR6 and postsynaptic density protein 95 (PSD95). Furthermore, these studies have shown that the activation of mixed lineage kinase 3 (MLK3), mitogen-activated kinase kinase 7 (MKK7) and c-Jun NH2-terminal kinase 3 (JNK3) are facilitated by kainate, resulting in neuronal cell death in the CA1 region (Tian et al., 2005).
Yang et al. (1997) have demonstrated that GluR6 knockout and JNK3-de fi cient mice exhibit similar phenotypes, and are resistant to excitotoxicity and kainate-induced seizures in the hippocampus. Our previous study has focused on Tat-GluR6-9c, a peptide containing the C terminus of GluR6 linked to the membrane transduction sequence Tat protein of HIV (Pei et al., 2006). This study has shown that the assembly of GluR6-PSD95-MLK3 signaling module is attenuated and protects neurons against cerebral ischemia/reperfusion-induced apoptosis. However, whether this signaling module-mediated JNK activation exists in the CA1 region of epileptic rats is still unknown. Therefore, in the presentstudy, we investigated whether recombinant adenovirus (Ad)-C-terminal amino acids of GluR6 (GluR6c) inhibited the assembly of the GluR6-PSD95-MLK3 signaling module and decreased kainate-induced neuronal death in the CA1 subregion.
Materials and Methods
Animals
A total of 24 adult male Sprague-Dawley rats, weighing 230 ± 20 g, were used and obtained from the Shanghai Experimental Animal Center, Chinese Academy of Science (Certi fi cate of Conformity Number 410116). All rats were housed in a laminar fl ow room at 18—22°C and a humidity of 55—58%. Drinking water and food were sterilized by steam. The experimental procedures were conducted according to the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China.
Establishment of seizure models
Seizures were induced by an intraperitoneal injection of kainate (12 mg/kg, dissolved in sterile saline). The rats were behaviorally monitored for seizures for at least 6 hours after injection. The seizures were scored using a modified scale (Racine, 1972): (1) behavioral arrest and staring spells, (2) head bobbing and gnawing, (3) unilateral forelimb clonus, (4) bilateral forelimb clonus, (5) severe seizures with loss of postural control, and (6) seizure-induced death. The rats that experienced epileptic seizures with stage 4 to 5 for more than three times were considered successful models. Only animals with stage 4 or 5 seizures were used in this study.
Sample preparation
The rats were decapitated at 3, 6, and 12 hours, and 1 and 3 days after kainate injection. The CA1 region was separated and quickly frozen in liquid nitrogen (Paxinos and Watson, 2007). The sample was homogenized in ice-cold homogenization buffer, supplemented with 50 mmol/L 3-(N-morpholino) propanesulfonic acid Sigma-Aldrich, St. Louis, MO, USA) (pH 7.4), 100 mmol/L KCl, 320 mmol/L sucrose, 50 mmol/L NaF, 0.5 mmol/L MgCl2, 0.2 mmol/L dithiothreitol, 1 mmol/L ethylenediamine tetraacetic acid, 1 mmol/L ethylene glycol tetraacetic acid, 1 mmol/L Na3VO4(Sigma-Aldrich), 20 mmol/L sodium pyrophosphate, 20 mmol/L β-phosphoglycerol, 1 mmol/L p-nitrophenyl phosphate, 1 mmol/L benzamidine, 1 mmol/L phenylmethylsulfonyl fl uoride, 5 μg/mL leupeptin, 5 μg/mL aprotinin, and 5 μg/mL pepstatin A. The homogenates were centrifuged at 800 × g at 4°C for 10 minutes. Supernatants were collected, and protein concentration was determined in accordance with a previous method (Lowry et al., 1951). Samples were stored at −80°C and were thawed only once for use.
Immunoprecipitation
Tissue homogenates (400 μg of protein) were diluted four-fold with 50 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer (pH 7.4), containing 10% glycerol, 150 mmol/L NaCl, 1% Triton X-100, 0.5% NP-40, and 1 mmol/L of ethylenediamine tetraacetic acid, ethylene glycol tetraacetic acid, phenylmethyl sulfonylfluoride and Na3VO4(all from Sigma-Aldrich). Samples were preincubated with 20 μL protein A sepharose CL-4B (Amersham, Uppsala, Sweden) for 1 hour at 4°C, and then centrifuged to remove proteins that adhered nonspeci fi cally to protein A. The supernatants were incubated with 1—2 μg of primary antibodies overnight at 4°C or for 4 hours. Protein A was added to the tube for an additional 2-hour incubation. Samples were centrifuged at 10,000 × g at 4°C for 2 minutes. The pellets were washed three times with immunoprecipitation buffer. Bound proteins were eluted by boiling in sodium dodecyl sulfate polyacrylamide gel electrophoresis loading buffer at 100°C for 5 minutes, and then isolated by centrifugation.
Western immunoblotting
Proteins extracted from CA1 supernatants were separated on polyacrylamide gels via electrophoresis, and then transferred to nitrocellulose membranes (Amersham Biosciences, Buckinghamshire, UK). After blocking with 3% serum albumin in Tris-buffered saline and 0.1% Tween-20 for 3 hours, membranes were incubated with mouse monoclonal anti-JNK antibody (1:1,000; Santa Cruz Biotechnology, Dallas, TX, USA), mouse monoclonal anti-p-JNK antibody (1:1,000; Santa Cruz Biotechnology), goat polyclonal anti-GluR6 (1:1,000; Santa Cruz Biotechnology), goat polyclonal anti-MKK7 (1:200; Santa Cruz Biotechnology), goat polyclonal anti-p-MKK7 (1:500; Cell Signaling, Boston, MA, USA), rabbit polyclonal anti-p-MLK3 (1:1,000; Cell Signaling), rabbit polyclonal anti-MLK3 antibody (1:200; Santa Cruz Biotechnology), or mouse monoclonal anti-PSD95 (1:1,000; Sigma-Aldrich) in Tris-buffered saline with 3% bovine serum albumin and Tween, overnight at 4°C. Rabbit polyclonal anti-Beta-actin (1:3,000; Santa Cruz Biotechnology) served as the housekeeping protein. Membranes were then washed and incubated with the secondary antibodies: goat anti-mouse (1:5,000; Sigma) or alkaline phosphatase-conjugated goat anti-rabbit (1:5,000; Sigma) in Tris-buffered saline with Tween at 25°C for 2 hours. Membranes were then developed with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate color substrate (Promega, Madison, WI, USA). The optical density of the protein bands (Target protein/ β-actin) on the membrane was scanned and analyzed by Lab Works image analysis software (UVP, Upland, CA, USA).
Histological analysis
The rats were perfusion-fixed with 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.4) under anesthesia, 7 days after kainate injection. Brains were removed quickly and further fixed in the same fixative at 4°C overnight. Post- fi xed brains were embedded in paraf fi n and sliced into 5-μm-thick coronal sections using a microtome (Leica, Wetzlar, Germany). Sections were dewaxed with xylene, rehydrated with ethanol at graded concentrations of 100—70% (v/v), and then washed with water. The sections were stainedwith 0.1% (w/v) cresyl violet and observed under the light microscope (Olympus, Tokyo, Japan). The number of surviving hippocampal CA1 pyramidal cells per 1-mm-length was counted as the neuronal density. Cells were counted on six random microscopic fi elds in a double-blind manner by two observers.
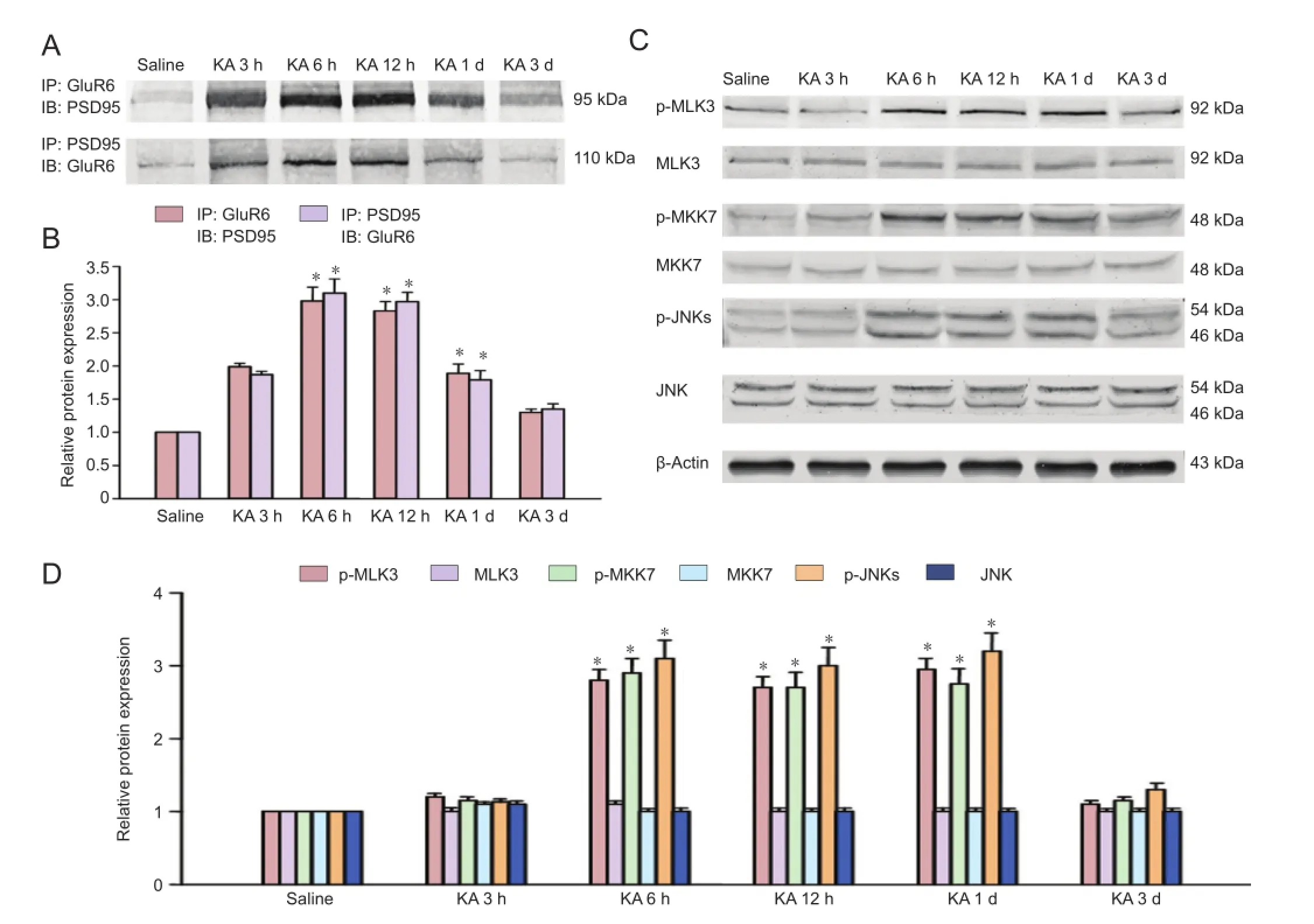
Figure 1 Time courses of the interactions of GluR6 with PSD95 and p-MLK3, p-MKK7 and p-JNKs derived from saline-treated rats or rats at various times of KA injection.
Recombination of adenoviral vectors
Recombinant Ad-GluR6c-green fluorescent protein constructs were produced in accordance with standard techniques (He et al., 1998). The pAd Track CMV vector is bicistronic, and expresses both green fl uorescent protein and the GluR6c domain. Briefly, GluR6c (852-908 amino acids of GluR6) was generated by polymerase chain reaction of the appropriate GluR6c coding region to incorporate lanking Bgl II and Hind III sites followed by ligation into the Ad shuttle vector pAdTrack-CMV digested with Bgl II and Hind III (Promega). The resultant plasmid was linearized by digestion with restriction endonuclease Pme I (New England Biolabs, Beverly, MA), and subsequently cotransformed into Escherichia coli (Promega). BJ5183 cells (Addgene, Cambridge, MA, USA) have an adenoviral backbone plasmid pAdEasy-1. Recombinants were selected with kanamycin, and recombination confirmed by restriction endonuclease analyses. Finally, the linearized recombinant plasmid was transfected into Ad packaging cell lines, Human Embryonic Kidney 293 cells (Addgene). Recombinant Ads were generated typically within 7 to 12 days, puri fi ed, and then tittered.
Drug treatment

Figure 2 Effect of pretreatment with adenovirus-GluR6c on the interactions of GluR6 with PSD-95 and the phosphorylation of MLK3, MKK7 and JNKs in the CA1 region in rats.
Rats were equally divided into saline , kainate-treated, Ad-treated and Ad-GluR6c groups. A single dose of kainate (12 mg/kg) was injected intraperitoneally to the rats, which were carefully monitored for signs of seizures. Within 15 minutes following the injection, rats first presented with deep breathing and increased salivation, followed by scratching, and then progression to rearing and generalized clonic/ tonic seizures within 50—60 minutes, which lasted for 2—3 hours. Two hours after the cessation of behavioral seizures, rats were taken back to their cages and sacri fi ced 7 days after the kainate injection. Control rats were only given 0.9% NaCl, the same volume of used for the kainate-treated rats. A total of 10 μL of Ad or Ad-GluR6c (1 × 1010pfu) was given to the rats of the Ad and Ad-GluR6c-treated groups 40 minutes before kainate injection to the CA1 region (anteroposterior: 3.6 mm; lateral: 2.0 mm; depth: 4.0 mm from bregma).
Statistical analysis
All data were expressed as the mean ± SD, and were analyzed by one-way analysis of variance followed by Duncan’s new multiple range method. Statistical analysis was performed using SPSS 13.0 software (SPSS, Chicago, IL, USA). A value of P < 0.05 was considered statistically signi fi cant.
Results
Alterations of the GluR6-PSD95-MLK3 signaling module during kainate-induced seizures in the CA1 region
Rats were injected with kainate for specific time-frames to explore the changes in the assembly of the GluR6-PSD95-MLK3 signaling module during seizures. Western immunoblotting were then performed for GluR6 or MLK3 with PSD95 at the speci fi c time points. The interactions of GluR6 and PSD95 following kainate injection increased rapidly, peaking at 6 hours and gradually decreasing to control levels 3 days later (Figure 1A, B). Saline did not affect the interactions of GluR6 and PSD95.
MLK3, an upstream kinase of MKK7 and JNK, can be activated by GluR6 and PSD95 (Savinainen et al., 2001). Therefore, we analyzed the effect of kainate on the activation (phosphorylation) of MLK3. Western immunoblotting revealed that kainate treatment increased the phosphorylation of MLK3 in the CA1 region (Figure 1C, D). The activation of JNK and MKK7 was signi fi cantly induced at 6 hours after kainate injection (P < 0.05).
Ad-GluR6-c suppressed kainate-induced activation of MLK3, MKK7, and JNK in the CA1 region
To elucidate whether downstream proteins of GluR6 was affected by the over-expression of GluR6c, Ad-GluR6c was administered to observe the variation of phosphorylated MLK3, MKK4/7 and JNKs (Figure 2A–D). Western immunoblotting revealed that Ad-GluR6c signi fi cantly (P < 0.05) inhibited the phosphorylation of MLK3 (Figure 2C, D). Additionally, the activation of MKK7 6 hours after kainate injection was significantly (P < 0.05) suppressed by Ad-GluR6c (Figure 2C, D). Similar results were obtained with JNKs (Figure 2C, D).
Neuroprotective effects of Ad-GluR6-c against kainateinduced neuronal injury in CA1 neurons
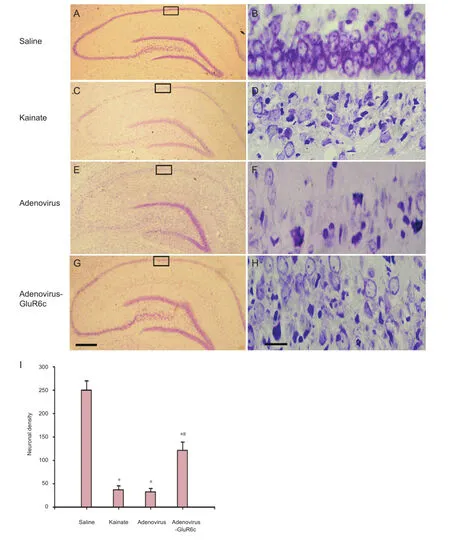
Figure 3 Neuroprotection of adenovirus-GluR6c against KA-induced brain damage in the CA1 region.
To investigate whether pretreatment with Ad-GluR6-c was protective against kainate-induced cell death, rats were pre-treated with Ad-GluR6c via a cerebroventricular injection 40 minutes before kainate administration. Rats from the saline, kainate, Ad, and Ad-GluR6c groups were perfusion- fi xed with paraformaldehyde, 7 days later. Cresyl violet staining was conducted to examine the survival of CA1 pyramidal cells. Our results showed normal CA1 neuronal cells as round and palely stained nuclei (Figure 3A, B), whereas kainate-induced cells showed pyknotic nuclei (Figure 3C, D), indicative of cell death. The pre-treatment of Ad-GluR6-c reduced neuronal degeneration (Figure 3G, H), whereas the Ad group did not show any protection against kainate-induced degeneration (Figure 3E, F). The neuronal densities of the saline, kainate, Ad and Ad-GluR6c groups were 250.0 ± 19.8, 37.2 ± 8.5, 32.6 ± 7.3, and 121.3 ± 17.8, respectively (Figure 3I).
Discussion
Many drugs have been developed for epilepsy in the past decades (Sander and Shorvon, 1996), but approximately one-third of epilepsy patients still cannot be cured. A larger percentage of patients suffer from the side effects of antiepileptic drugs (Smith and Bleck, 1991). We showed that in the CA1 region, Ad-GluR6c inhibited the 6-hour kainate-induced activation of MLK3, MKK7, and JNK. Furthermore, pretreatment with Ad-GluR6c significantly protected neuronal cells in the CA1 region from kainate-induced death. Overall, these results suggest that Ad-GluR6c generates the GluR6c peptide in neuronal cells and possibly binding to the PDZ1 domain of PSD95, then suppressing the interaction of PSD95 and GluR6.
Administration of kainate has been shown to increase mitochondrial dysfunction, induce the production of reactive oxygen species, and induce apoptosis in many regions of the brain, particularly in the CA1 region (Wang et al., 2005; Guo et al., 2012; Yuan etal., 2014). Kainate-induced neuronal injury in the hippocampus is reversed by the activation of adenosine A receptors (Matsuoka et al., 1999), dopamine D2 receptors (Bozzi et al., 2000), and N-methyl-D-aspartate receptors (Ogita et al., 2003). The activation of the kainate receptor subunit GluR6 induces neuronal cell death in the hippocampus (Liu et al., 2006). Moreover, GluR6 knockout mice have shown resistance to neuronal cell death and to kainate-induced seizures (Mulle et al., 1998). However, the precise molecular mechanism underlying the effect of GluR6 remains unclear. Savinainen et al. (2001) have reported that GluR6, MLK3, and PSD95 form a signaling complex and facilitate the activation and phosphorylation of MLK3 and JNK in vitro. In the present study, we demonstrated suppressing the assembly of the GluR6-PSD95-MLK3 signaling module attenuated MLK3 and JNK activation and kainate-induced seizures in vivo.
Members of MLK regulate the JNK signaling pathway by phosphorylation-dependent regulation of MKK4 and MKK7 (Muniyappa and Das, 2008; Wen et al., 2008; Mishra et al., 2010; Wang et al., 2011; Chen et al., 2012; Chen and Gallo, 2012; Song et al., 2012; Wang and Xia, 2012; Zhang et al., 2012; He et al., 2013; Owen et al., 2013; Rana et al., 2013). MKK4 and MKK7 are dual-speci fi city kinases phosphorylating threonine and tyrosine residues in the catalytic domains of JNK (Davis, 2000). Numerous studies have demonstrated that the JNK signaling pathway plays an important role in mediating neurotoxicity (Saporito et al., 1998; Behrens et al., 1999; Wu et al., 2000; Borsello et al., 2003; Kuan et al., 2003; Zhang et al., 2006; Moon et al., 2013; Oshitari et al., 2013; Chen et al., 2014; Lu et al., 2014). The MLK-MKK7-JNK signaling module has been shown to be regulated by the activation of JNK3 (Whitmarsh et al., 1998), which is involved in kainate-induced brain injury (Liu et al., 2006). Our previous study has clearly demonstrated the activation of JNK3 and its association with neuronal cell death during brain ischemia/reperfusion (Tian et al., 2003). Our present results showed that application of Ad-GluR6c inhibited the assembly of the GluR6-PSD95-MLK3 signaling module, and subsequently attenuated the activation of MLK3 and JNK.
In summary, kainate induced the assembly of the GluR6-PSD95-MLK3 signaling module, and subsequently activated the downstream JNK signaling pathway, ultimately resulting in neuronal cell death. Application of Ad-GluR6c suppressed the binding of GluR6 to the PDZ1 domain of PSD95 in the postsynaptic regions, and subsequently inhibited the assembly of the GluR6-PSD95-MLK3 signaling module by inhibiting the activation of MLK3 and JNK.
Author contributions:Mou J and Liu XM provided study data, ensured the integrity of the data, participated in data analysis, and wrote the manuscript. Mou J participated in study concept and design. Pei DS was in charge of manuscript authorization, provided technical or material support, obtained the funding and served as a principle investigator. All authors approved the final version of this paper.
Con fl icts of interest:None declared.
Behrens A, Sibilia M, Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21:326-329.
Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9:1180-1186.
Bozzi Y, Vallone D, Borrelli E (2000) Neuroprotective role of dopamine against hippocampal cell death. J Neurosci 20:8643-8649.
Bureau I, Bischoff S, Heinemann SF, Mulle C (1999) Kainate receptor-mediated responses in the CA1 fi eld of wild-type and gluR6-defi cient Mice. J Neurosci 19:653-663.
Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS, Wang HL (2012) (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ 19:1623-1633.
Chen J, Gallo KA (2012) MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res 72:4130-4140.
Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q, Guo M, Huang S, Chen L (2014) Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J Neurochem 128:256-266.
Darstein M, Petralia RS, Swanson GT, Wenthold RJ, Heinemann SF (2003) Distribution of kainate receptor subunits at hippocampal mossy fi ber synapses. J Neurosci 23:8013-8019.
本发明公开了一种化学镀铜溶液用安定剂,由下述质量份原料组成:三水合亚铁氰化钾22 ~ 26 g/L,四水合酒石酸钾钠60 ~ 70 g/L,促进剂2-硫醇基苯骈噻唑0.38 ~ 0.39 g/L,水1 L。化学镀铜溶液用安定剂的制备方法,具体步骤如下:常温下,向水中加入三水合亚铁氰化钾,并搅拌5 ~ 10 min,然后加入四水合酒石酸钾钠,搅拌5 ~ 10 min,最后加入2-硫醇基苯骈噻唑,搅拌至完全溶解。本发明具有能够提高化学铜镀液的稳定性,减少和避免铜离子歧化的优点。
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239-252.
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7-62.
Guo DH, Liu XH, Zeng J, Tang Y, Zeng WJ, Luo ZZ, Lei YL, Yu HX (2012) Effect of the re-distribution of kainate 1 expression on the neuronal excitotoxicity. Zhongguo Zuzhi Gongcheng Yanjiu 16:287-290.
He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z, Xiao Y (2013) miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/ c-jun pathway. Biochem Biophys Res Commun 441:763-769.
He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509-2514.
Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P (2003) A critical role of neural-speci fi c JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A 100:15184-15189.
Liu XM, Pei DS, Guan QH, Sun YF, Wang XT, Zhang QX, Zhang GY (2006) Neuroprotection of Tat-GluR6-9c against neuronal death induced by kainate in rat hippocampus via nuclear and non-nuclear pathways. J Biol Chem 281:17432-17445.
Lu TH, Tseng TJ, Su CC, Tang FC, Yen CC, Liu YY, Yang CY, Wu CC, Chen KL, Hung DZ, Chen YW (2014) Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol Lett 224:130-140.
Matsuoka Y, Okazaki M, Takata K, Kitamura Y, Ohta S, Sekino Y, Taniguchi T (1999) Endogenous adenosine protects CA1 neurons from kainic acid-induced neuronal cell loss in the rat hippocampus. Eur J Neurosci 3617-3625.
Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B (2010) Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol 24:598-607.
Moon MH, Jeong JK, Lee YJ, Park SY (2013) FTY720 protects neuronal cells from damage induced by human prion protein by inactivating the JNK pathway. Inter J Mol Med 32:1387-1393.
Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-de fi cient mice. Nature 392:601-605.
Muniyappa H, Das KC (2008) Activation of c-Jun N-terminal kinase (JNK) by widely used speci fi c p38 MAPK inhibitors SB202190 and SB203580: a MLK-3-MKK7-dependent mechanism. Cell Signal 20:675-683.
Ogita K, Okuda H, Yamamoto Y, Nishiyama N, Yoneda Y (2003) In vivo neuroprotective role of NMDA receptors against kainate-induced excitotoxicity in murine hippocampal pyramidal neurons. J Neurochem 85:1336-1346.
Oshitari T, Bikbova G, Yamamoto S (2013) Increased expression of phosphorylated c-Jun and phosphorylated c-Jun N-terminal kinase associated with neuronal cell death in diabetic and high glucose exposed rat retinas. Brain Res Bull 101C:18-25.
Owen GR, Achilonu I, Dirr HW (2013) High yield purification of JNK1beta1 and activation by in vitro reconstitution of the MEKK1-->MKK4-->JNK MAPK phosphorylation cascade. Protein Expr Purif 87:87-99.
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates, 6thEdition. Amsterdam, Boston: Academic Press, Elsevier, USA.
Pei DS, Wang XT, Liu Y, Sun YF, Guan QH, Wang W, Yan JZ, Zong YY, Xu TL, Zhang GY (2006) Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain 129:465-479.
Racine RJ (1972) Modi fi cation of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophys 32:281-294.
Rana A, Rana B, Mishra R, Sondarva G, Rangasamy V, Das S, Viswakarma N, Kanthasamy A (2013) Mixed lineage kinase-c-Jun N-terminal kinase axis: a potential therapeutic target in cancer. Genes Cancer 4:334-341.
Sander JW, Shorvon SD (1996) Epidemiology of the epilepsies. J Neurol Neurosurg Psychiatry 61:433-443.
Saporito MS, Brown ER, Carswell S, Di Camillo AM, Miller MS, Murakata C, Neff NT, Vaught JL, Haun FA (1998) Preservation of cholinergic activity and prevention of neuron death by CEP-1347/ KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience 86:461-472.
Savinainen A, Garcia EP, Dorow D, Marshall J, Liu YF (2001) Kainate receptor activation induces mixed lineage kinase-mediated cellular signaling cascades via post-synaptic density protein 95. J Biol Chem 276:11382-11386.
Smith MC, Bleck TP (1991) Convulsive disorders: toxicity of anticonvulsants. Clin Neuropharmacol 14:97-115.
Song YJ, Zong ZM, Liu HZ, Mukasa R, Pei DS, Mou J, Wen XR, Liu ZA, Wei XY (2012) Heme oxygenase-1 regulates the JNK signaling pathway through the MLK3-MKK7-JNK3 signaling module in brain ischemia injury. Brain Res 1429:1-8.
Tian H, Zhang G, Li H, Zhang Q (2003) Antioxidant NAC and AMPA/ KA receptor antagonist DNQX inhibited JNK3 activation following global ischemia in rat hippocampus. Neurosci Res 46:191-197.
Tian H, Zhang QG, Zhu GX, Pei DS, Guan QH, Zhang GY (2005) Activation of c-Jun NH2-terminal kinase 3 is mediated by the GluR6. PSD-95.MLK3 signaling module following cerebral ischemia in rat hippocampus. Brain Res 1061:57-66.
Wang J, Xia Y (2012) Assessing developmental roles of MKK4 and MKK7 in vitro. Commun Integr Biol 5:319-324.
Wang Q, Yin XH, Liu Y, Zhang GY (2011) K252a suppresses neuronal cells apoptosis through inhibiting the translocation of Bax to mitochondria induced by the MLK3/JNK signaling after transient global brain ischemia in rat hippocampal CA1 subregion. J Recept Signal Transduct Res 31:307-313.
Wang Q, Yu S, Simonyi A, Sun GY, Sun AY (2005) Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 31:3-16.
Wen XR, Li C, Zong YY, Yu CZ, Xu J, Han D, Zhang GY (2008) Dual inhibitory roles of geldanamycin on the c-Jun NH2-terminal kinase 3 signal pathway through suppressing the expression of mixed-lineage kinase 3 and attenuating the activation of apoptosis signal-regulating kinase 1 via facilitating the activation of Akt in ischemic brain injury. Neuroscience 156:483-497.
Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281:1671-1674.
Wu DC, Ye W, Che XM, Yang GY (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab 20:1320-1330.
Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865-870.
Yuan L, Zhang HX, Qian SL, Xu B, Gong JQ, Liu XH, Tang Y, Yu HX (2014) Kainic acid-induced endoplasmic reticulum stress model. Zhongguo Zuzhi Gongcheng Yanjiu 18:5861-5867.
Zhang QX, Pei DS, Guan QH, Sun YF, Liu XM, Zhang GY (2006) Blockade of the translocation and activation of mitogen-activated protein kinase kinase 4 (MKK4) signaling attenuates neuronal damage during later ischemia-reperfusion. J Neurochem 98:170-179.
Zhang Y, Li F, Liu S, Wang H, Mahavadi S, Murthy KS, Khalili K, Hu W (2012) MEKK1-MKK4-JNK-AP1 pathway negatively regulates Rgs4 expression in colonic smooth muscle cells. PLoS One 7:e35646.
Copyedited by Mark F, Wysong S, Wang J, Qiu Y, Li CH, Song LP, Zhao M
10.4103/1673-5374.147932
Dongsheng Pei, Ph.D., Jiangsu Key Laboratory of Biological Cancer Therapy, Xuzhou Medical College, Xuzhou 221002, Jiangsu Province, China, dspei@xzmc.edu.cn.
http://www.nrronline.org/
Accepted: 2014-09-22
猜你喜欢
杂志排行

中国神经再生研究(英文版)的其它文章
- Angioplasty and stenting for severe vertebral artery ori fi ce stenosis: effects on cerebellar function remodeling veri fi ed by blood oxygen level-dependent functional magnetic resonance imaging
- A more consistent intraluminal rhesus monkey model of ischemic stroke
- Human bone marrow mesenchymal stem cell transplantation attenuates axonal injury in stroke rats
- Pathogenesis of glaucoma: how to prevent ganglion cell from axonal destruction?
- Puerarin protects brain tissue against cerebral ischemia/reperfusion injury by inhibiting the in fl ammatory response
- Pretreatment with scutellaria baicalensis stem-leaf total fl avonoid protects against cerebral ischemia/ reperfusion injury in hippocampal neurons