Pathogenesis of glaucoma: how to prevent ganglion cell from axonal destruction?
2014-06-01FranciscoJavierCarreras
Pathogenesis of glaucoma: how to prevent ganglion cell from axonal destruction?
Glaucoma is the main cause of irreversible blindness globally, but the pathogenesis of glaucoma is still pending consensus. In spite of great advances during the 20thand early 21stcenturies in terms of detailed knowledge of pathological processes that take place in glaucoma, the exact pathogenesis is much less clear. Hypotheses abound, and the most researched areas are ischemia at the optic nerve head, blockade of ganglion cell axonal transport, peripapillary atrophy, and changes in the characteristics of the lamina cribrosa (Nucci et al., 2008).
Although it is a well-established clinical fact that very high intraocular pressure (IOP), higher than blood perfusion pressure, is able to induce ischemic damage, the presence of glaucomatous changes in patients with moderately increased and normal or even low pressure, without demonstrable pressure spikes, cannot directly be attributed to pressure, and additional explanations have been sought. Rather than one pathological process, the clinical recognition of low-pressure glaucoma has brought about complementary explanations, creating different categories of glaucoma with different pathogenesis.
Raised intraocular pressure has been dropped as “the” causative factor and now ranks alongside other risk factors. As the number of known risk factors multiplies, complex mathematical approaches are deemed necessary to tackle the tangled relationships between risk factors. Lowering IOP continues to be the only modi fi able risk factor, and the mainstay of glaucoma treatment. But the progression of open angle glaucoma (OAG) in many cases, despite IOP treatment, strongly suggests that other factors must play a part in the development of the disease. Although dissatisfaction with a single approach based on ocular hypertension has helped promote direct anti-ischemic and neuroregenerative therapeutic approaches, these have been largely abandoned as sterile, when not outright dangerous (Caprioli et al. 2010).
With a wealth of data at hand, there is an apparent urgent need to seek new causal relationships between known deviations from the normal physiology both at the organ level and cell level, and propose, as a working hypothesis, a new pathogenetic model, simple, congruent with the clinical heritage, and with feasible therapeutic applications. But is it possible?
Focused on basic research on the pathogenesis of glaucoma, we have been working in our laboratory with cultured optic nerve tissue. Our approach to the pathology of glaucoma is condensed in a proposition based on histological evidence and presented as a chain of events. In short, we contemplate glaucoma as a result of the misdirection of the aqueous out fl ow.
The new hypothesis tries to solve some salient inconsistencies that form part of the narrative of the “hypertensive- mechanical-ischemic paradigm”. Only some of those inconsistencies, and their proposed solutions, will be considered here. It is frequent to fi nd in the specialized literature that in the characterization of the pathology of glaucoma only the retinal ganglion cells (RGCs) and their axons are mentioned. Astrocytes are either plainly ignored, or even, in some cases, described as “reactive astrocytes” with newly arisen lethal abilities that would damage rather than protect the axons that depend on them. Other aspects that are dif fi cult to interpret at the ultrastructural level are how a process that causes progressive damage over decades, and projects a consistent diagnostic pattern in the visual fi elds is produced by pressure oscillation2s in a single, not lobulated, capillary bed in an area of 2.5 mm, part of a vascular bed with autoregulatory capabilities. As a majority of glaucoma cases fall into the categories of moderate and low tension glaucoma, zeal to uncover pathological mechanisms in the hypertensive- mechanical-ischemic damage has led to dif fi culties to explain why normal individuals do not have glaucoma, or to dilute OAG in the murky waters of more complex syndromes, as the vascular dysregulation syndrome (Mozaffarieh and Flammer, 2009).
We demanded our hypothesis to face the following principles: in order to be considered, the hypothesis should be able to explain loss of axons under hypertensive and hypotensive situations. Axon loss should be able to occur in a very progressive manner, either slow or accelerated. It should be a process capable of leading to total blindness and also able to stop after some nerve loss, and either not progressing further or significantly slowing the rate of loss. At least the initial damage should take place in the optic nerve head (ONH) and should be based on the proven fact that the out fl ow of aqueous through the conventional and alternative pathways is altered during glaucoma development.
I will set out here a few structural details of the optic nerve head histological architecture that helped to shape our proposition. The surface of the vitreous surface of the optic nerve is formed by astrocytic projections. Around the main vessels there is a net of wide interconnected intercellular spaces; those passages are absent in the areas only occupied by axons and axon-wrapping astrocytes. No tight junctions are present connecting the astrocytic projections, only occasional adherens and gap junctions are present (Carreras et al., 2009). Excess extracellular fluid can be interchanged with the vitreous by the network of interconnected peri-vascular and para-axonal spaces (Carreras et al., 2010a). That the vitreous body itself is permeable in mammals to fl uids and solutes has been known since the late 19thcentury. It means there is no obstacle to aqueous humor diverted to the vitreous cavity being evacuated though the optic nerve head (Carreras et al., 2010b). As Figure 1A shows, in case of increased resistance to out fl ow through the anterior pathways, the natural exit for aqueous is the optic nerve head. The colloidosmotic pressure of the vitreous body, the only obstacle to posterior outfl ow, is variable and changes with age.
Modeling of the retina and optic nerve has shown that following a model of development of the optic stalk, in which order of axonal grow and fasciculation is strictly followed, reproduces the path of the axons in the retina with up to 90% fi delity (Carreras et al., 2011). These models show the existence of a singularity for the fi bers coming temporarily to the macula that enter the optic disc centrally rather than peripherally. If a concentric erosion of the optic cup is implemented, then the early visual defect of nasal step is reproduced (Carreras et al., 2010b). As a diagramatic depiction of the ordering of the axons in the neuroretinal rim shows (Figure 1B), antero-posterior elimination of axons is coincident with an enlarging central cup. Both could be the result of a noxa (the aqueous humor) acting from front to rear. The mentioned singularity would explain the early appearance of the nasal step in glaucomatous fi elds (Carreras et al., 2011a).
The nutrition of RGCs, axons depends on the transport across the astrocytes of lactate and, in lesser measure, glucose. Metabolic coupled molecules, monocarboxylate transporters (MCTs) and glucose transporters (GLUTs), need the presence of gap junctions along with CAMs and adherens junctions to keep the energy shuttle in place (Carreras et al. 2011b). Figure1C shows the elements that participate in the energy shuttle from the capillaries to the axons. Key elements are represented: gap junctions, adherens junctions, MCTs and GLUTs that cooperate in the metabolic energy transfer.
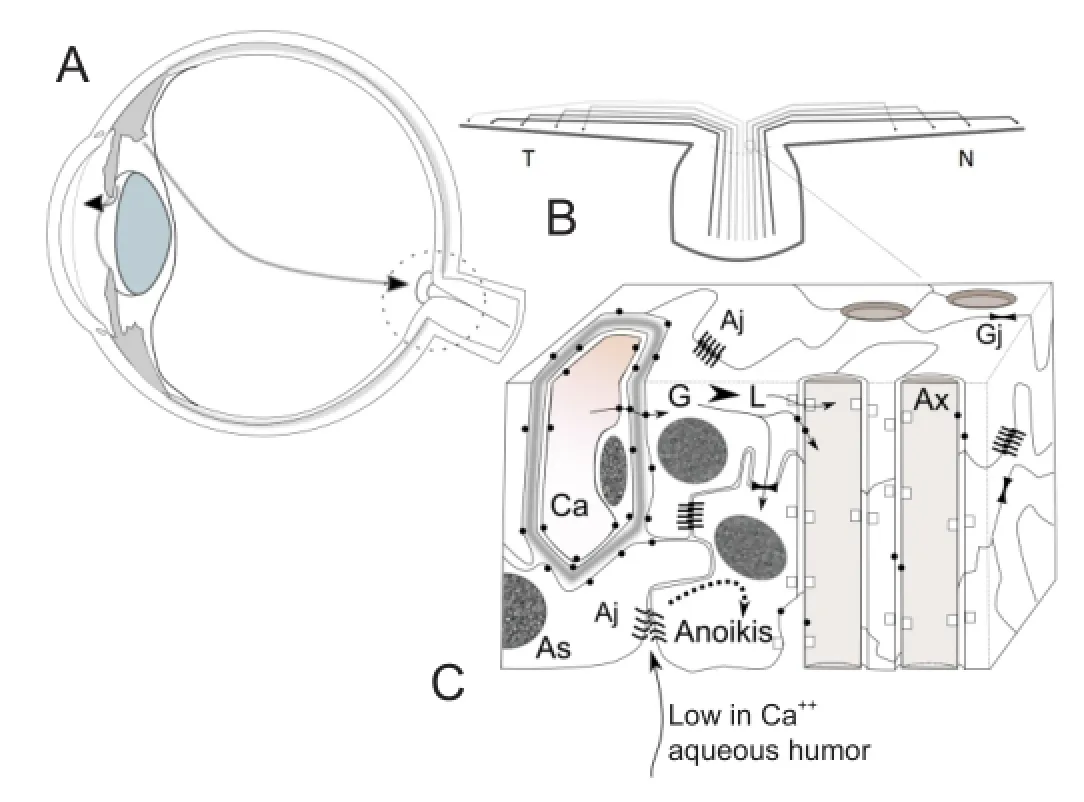
Figure 1 Misdirection of aqueous out fl ow in glaucoma and ionic stress at the optic nerve head level.
The previous structural data gain relevance when the ionic composition of the aqueous humor is recalled. Ionic composition of aqueous humor is critically different from that of the plasma and intersititial tissue. Both in vitreous and aqueous, the concentration of ion calcium is lower than in plasma, varying from 49% to 57% of plasma levels, while the concentration of potassium ion is similar to that of plasma (Carreras et al., 2009). This combination is crucial to adherens junctions, which require calcium to establish homophilic cadherin interactions, and potassium, a smaller cation, interferes with the union. The levels of calcium and potassium present in the aqueous have been shown to trigger a salvo of apotosis in prelaminar tissue culture (Carreras et al., unpublished results). Due to their signalling role, separation of adherens junctions by a drop in calcium concentration is able to trigger a form of apoptosis known as anoikis. The axons falls secondarily to astrocyte support deprivation. Axonal compartimentalized destruction is followed secondarily by somatic RGC apoptosis. Anteroposterior thinning of the fi ber layer and erosion of the central cup would ensue, giving rise to the characteristic glaucoma progression in the visual fi elds. This process is summarized in Figure 1C. This process of “silent” tissue destruction would explain, among others signs, splinter disc hemorrhages as a consequence of capillary breakdowns due to lack of tissue support.
In brief, this hypothesis blames not the raised intraocular pressure, but the misdirection of the fl ow of aqueous towards the posterior pole of the eye as the main culprit in glaucoma. If proven, this might have huge consequences both in the diagnosis of the disease and follow-up and treatment. From this perspective, the hypothesis would suggest interfering with the axonal destruction process, that could be prevented by avoiding the posterior fl ow of aqueous humor through the optic nerve. This view opens interesting perspectives about the role that the preservation of the integrity and colloidosmotic properties of the vitreous body might have in the prevention of this blinding and widespread disease.
The crucible of this proposition will be the clinical tests to relate quantitatively posterior fl ow of aqueous and glaucoma progression, and for that purpose the adaptation of new optic technology would be required. If successful, new clinical tests could advance the diagnosis of glaucoma to precede severe fi ber loss.
Francisco Javier Carreras
Department of Surgery (Ophthalmology), Faculty of Medicine, University of Granada, Avda. de Madrid, s/n - 18071-Granada, Spain
Caprioli J, Coleman AL (2010) Blood flow in glaucoma discussion. Blood pressure, perfusion pressure, and glaucoma. Am J Ophthalmol 149:704-712.
Carreras FJ, Porcel D, Garzón I, Alaminos M (2009) Cell-cell adhesion in the prelaminar region of the optic nerve head: a possible target for ionic stress. Ophthalmic Res 42:106-111.
Carreras FJ, Porcel D, Muñoz-Avilés I (2010a) Mapping the surface astrocytes of the optic disc: a fl uid-conducting role of the astrocytic covering of the central vessels. Clin Experiment Ophthalmol 38:300-308.
Carreras FJ, Porcel D, Guerra I, Carreras I (2010b) Fenestrations and preferential flow routes in the prelaminar optic nerve through wet scanning electron microscope and perfusion of tracers. Clin Experiment Ophthalmol 38:705-717.
Carreras FJ, Rica R, Delgado AV (2011a) Modeling the patterns of visual fi eld loss in glaucoma. Optom Vis Sci 8:E63-79.
Carreras FJ, Porcel D, Rodriguez-Hurtado F, Zarzuelo A, Carreras I, Galisteo M (2011b) Expression of Metabolic Coupling and Adhesion Proteins in the Porcine Optic Nerve Head: Relevance to a Flow Model of Glaucoma. Chapter 4 in The Mistery of Glaucoma. Dr. Tomaš Kubena (Editor)- InTech Open Access Publisher. ISBN 978-953-307-567-9.
Mozaffarieh M, Flammer J (2009) Ocular blood fl ow and glaucomatous optic neuropathy. Springer Verlag, Berlin Germany
Nucci C, Osborne N, Bagetta G, Cerulli L (2008) Glaucoma: an open-window to neurodegeneration and neuroprotection. Elsevier, Burlington, MA, USA.
Francisco Javier Carreras, M.D., Ph.D.
Email: fcarrera@ugr.es.
10.4103/1673-5374.147928 http://www.nrronline.org/
Accepted: 2014-11-17
Carreras FJ. Pathogenesis of glaucoma: how to prevent ganglion cell from axonal destruction? Neural Regen Res. 2014;9(23):2046-2047.
杂志排行

中国神经再生研究(英文版)的其它文章
- Angioplasty and stenting for severe vertebral artery ori fi ce stenosis: effects on cerebellar function remodeling veri fi ed by blood oxygen level-dependent functional magnetic resonance imaging
- A more consistent intraluminal rhesus monkey model of ischemic stroke
- Human bone marrow mesenchymal stem cell transplantation attenuates axonal injury in stroke rats
- Puerarin protects brain tissue against cerebral ischemia/reperfusion injury by inhibiting the in fl ammatory response
- Pretreatment with scutellaria baicalensis stem-leaf total fl avonoid protects against cerebral ischemia/ reperfusion injury in hippocampal neurons
- Overexpression of C-terminal fragment of glutamate receptor 6 prevents neuronal injury in kainate-induced seizure via disassembly of GluR6-PSD95-MLK3 signaling module