同位素标记相对和绝对定量蛋白组技术研究进展
2014-05-04孔汉金张克山刘永杰尚佑军吴斌刘湘涛
孔汉金,张克山,刘永杰,尚佑军,吴斌,刘湘涛
1.中国农业科学院 兰州兽医研究所,家畜疫病病原生物学国家重点实验室,国家口蹄疫参考实验室,甘肃 兰州730046;2.华中农业大学 农业微生物学国家重点实验室,动物医学院,湖北 武汉 430070
蛋白质组学是一种新兴的高通量分析技术,与基因组学方法相结合,不仅能深入分析单个蛋白,还能对总体蛋白表达谱进行分析,能在总体层面上阐释蛋白-蛋白相互作用以及细胞生物学事件,有助于发现各细胞因子之间全新的联系。随着蛋白质组学研究的深入,人们已不再满足对生物个体组织细胞蛋白质组进行定性研究,而是着眼于蛋白质量的研究。定量蛋白质组学就是把一个基因组表达的全部蛋白质或一个复杂的混合体系中所有的蛋白质进行精确的定量和鉴定,在比较蛋白质组学研究中扮演重要角色。通过比较正常与异常细胞或组织中蛋白质表达水平的差异,找到与疾病密切相关的差异蛋白,进而确定靶分子,为临床诊断、病理研究、药物筛选、新药开发、新陈代谢研究等提供理论依据。但这些蛋白很少处于有或没有的状态,而是更多地呈现不同丰度的水平,因此必须采用一种无偏见的策略,以一种敏感和准确的方法衡量这些蛋白的变化。鸟枪法蛋白组学策略能够有效鉴定不同细胞和组织特定状态下上调和下调的蛋白。基于稳定同位素标签和液相色谱与串联质谱,最早由Gygi[1]等提出的同位素亲和标记(isotope-codedaffinity tags,ICAT)技术,其最大的缺点是蛋白质序列中必须含有半胱氨酸,那些在功能上有重要作用但不含半胱氨酸的蛋白质将会被遗失。美国应用生物系统公司(Applied Bio⁃systems Incorporation,ABI)研发的功能强大的同位素标记相对和绝对定量技术(isobaric tags for rela⁃tive and absolute quantification,iTRAQ)能在同一实验中同时对8种不同样品进行定性和定量分析,具有很好的精确性和重复性,并且弥补了ICAT技术的不足,迅速被广大学者接受。我们简要综述iTRAQ在定量蛋白组史中的地位作用、研究策略,及其在病毒致病机制研究和医药临床相关问题中的应用。
1 定量蛋白组学的研究历史
过去20年,大量基因组数据信息的出现彻底改变了利用化学方法鉴定单个蛋白的方式。蛋白数据库中覆盖众多蛋白,并且增加了注释部分,使蛋白信息更详细。但是数据库中的蛋白信息只能用于定性鉴定,目前还不可能仅根据数据库中的参比蛋白对摄动系统内蛋白表达水平进行差异比较,不过结合胶染色和蛋白标签技术去探知不同系统中蛋白表达的平行比较还是可能的。
传统双向电泳通过凝胶分离和质谱鉴定差异表达蛋白存在许多局限性,如不能检测具有极端等电点的、分子过大或过小的、低丰度的蛋白及膜蛋白,而且胶依赖技术的重现性较低,意味着定量困难和不准确。涉及同位素标签的鸟枪法蛋白组技术克服了传统双向电泳策略定量的诸多问题[2]。细胞培养条件下稳定同位素标记技术(stable isotope label⁃ing with amino acids in cell culture,SILAC)采用含有轻、重同位素型必需氨基酸的培养基进行细胞培养,使胞内蛋白被同位素稳定标记,将蛋白质等量混合后进行分离和质谱鉴定,氨基酸代谢渗入蛋白中,导致相应肽的质量偏移和质谱峰值强度改变,从而显示蛋白的相对丰度,对不同样品间的相同蛋白做出相对定量,属于体内代谢标记法[3]。尽管SILAC是一种高通量、高灵敏度、标记效果稳定的技术,但其主要缺陷在于必须依赖细胞系的内源标签,因而只能用于活体培养的细胞或低等有机体(如蠕虫),对于疾病研究中常用的组织样品、体液样品等无法分析。同位素亲和标签技术(isotope affinity coded tag,ICAT)是一种能比较2个不同样品中含半胱氨酸残基的特异性蛋白的标签策略[1]。2组样品分别通过重链和轻链标记后混合,经液相色谱分离质谱鉴定差异表达蛋白。ICAT标记的肽段信号强度定量着2种样品中同一蛋白的相对丰度。ICAT技术减少了样品的复杂性,但其对半胱氨酸残基的特异性意味着不含半胱氨酸的肽段将不被标记,那些在功能上有重要作用但不含半胱氨酸的蛋白将会被遗失。为了克服ICAT技术的主要局限,ABI公司开发了iTRAQ技术,这是一种新的、功能强大的进行绝对和相对定量研究的方法[4]。该技术起初的设计可同时对4组样品进行分析,现已发展到8组样品[5]。iTRAQ试剂为肽段N端及赖氨酸侧链氨基标记同重元素,标记后的不同样本间的同一蛋白表现为相同的质荷比,意味着不同样品的同一肽段在一级质谱中表现为同一信号峰,减少了样品分析的复杂性。而在二级质谱中,肽段丢失平衡基团后信号离子表现为不同质荷比的峰,根据波峰的高度及面积可以定量不同样品间的同一蛋白。
2 iTRAQ试剂结构
iTRAQ试剂是一种小分子同重元素化学物质,包括报告基团、肽反应基团和平衡基团,组合成相对分子质量为145的基团(图1)。iTRAQ试剂通过肽反应基团与肽段N端和赖氨酸侧链上的氨基结合,几乎可以标记样本中的所有蛋白。标记肽段经MS/MS打碎后,报告基团脱落产生不同质荷比(114、115、116、117、118、119、121和122)的诊断离子。诊断离子是iTRAQ技术蛋白质定量研究的关键,其峰高及面积与样品中同一肽段的相对丰度呈正比。iTRAQ标记肽除了能产生用于肽段定量的诊断离子外,还产生强大的y-ion和b-ion离子信号用于蛋白鉴定。iTRAQ标签设计中,选择合适的报告基团可减少低质量区域如基质、碎片离子噪音的干扰。iTRAQ 8-plex试剂报告基团缺少120,因为苯丙氨酸质荷比为120,会对质谱鉴定结果有干扰。
3 iTRAQ的基本实验流程
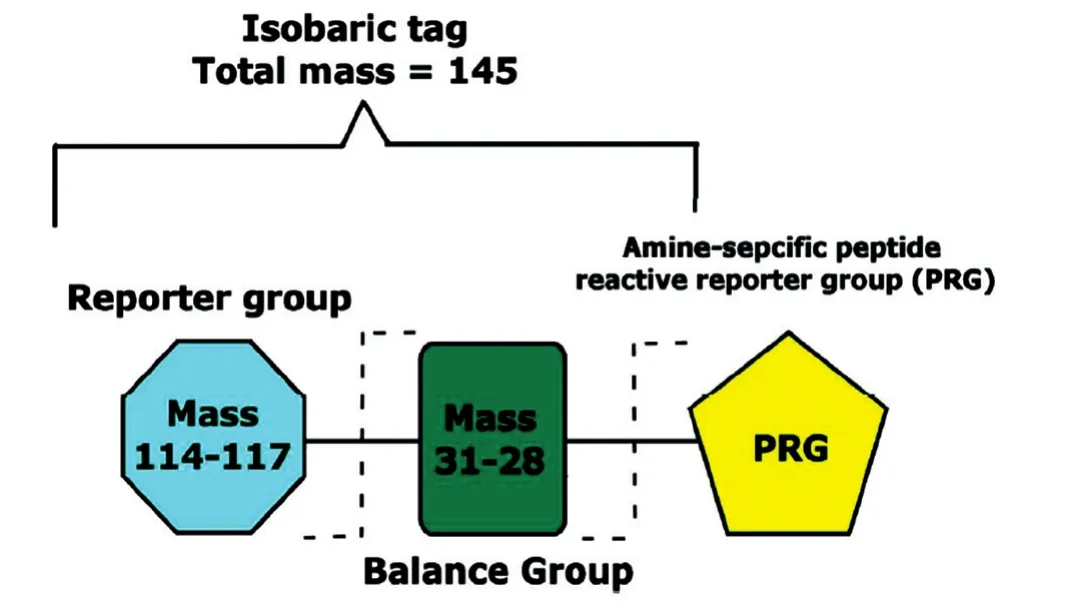
图1 iTRAQ试剂结构图[6]
iTRAQ实验流程包括样品还原、烷基化、半胱氨酸封闭和胰蛋白酶消化。消化后的肽段用4或8-plex iTRAQ试剂标记,然后混合到同一支试管内,经色谱纯化后进行质谱检测及分析。质谱分析中,不同样品来源的相同肽段以单一峰出现(iTRAQ平衡基团确保所有标记呈现相同的质荷比)。在二级串联质谱,iTRAQ报告基团脱落,其相对强度用于定量。每个四肽段以相同方式产生b-ion和y-ion离子用于蛋白鉴定。一般情况下iTRAQ并不需要在酶解前分离蛋白,但为了检测低丰度蛋白,可提前对样品进行分离,富集感兴趣的蛋白组分。经一维液相分离的肽段上质谱鉴定会存在高丰度肽段掩盖低丰度蛋白的检测,目前往往偶合多维分离蛋白鉴定技术(MudPIT)[7],包括亲和层析、离子交换层析、反向色谱层析和凝胶排阻层析。质谱检测后的数据经由生物信息学元件分析用于蛋白定量和鉴定。已经开发了多种功能强大的用于iTRAQ数据库搜索和分析的生物信息学元件,其中MASCOT是目前应用最为广泛的蛋白搜索引擎。这些生物信息软件具有很多特性,包括排除二级质谱iTRAQ报告基团、肽段N端修饰、赖氨酸修饰、半胱氨酸残基的甲基硫甲磺酸酯(MMTS)修饰的光谱鉴定及校正iTRAQ报告基团的肽谱峰值。而且这些生物信息分析软件也可自动完成相对定量。GPS Explorer(AB Sciex)能够定量数据库中所有肽段的iTRAQ蛋白和肽段比值。选择一个标签作为基准质量,比值通过片段校正面积/参考校正面积得到。计算过程中常常用到的归一化因子能够规范不同样品因含不平等总蛋白导致的iTRAQ比值异常。
4 iTRAQ实验设备和定量的准确性
iTRAQ技术中两大最普遍的质谱仪分别为MALDI-TOF-MS/MS和ESI-TOF-MS/MS,两者在不同iTRAQ分析中的准确性和性能各有所长。Kuzyk和Scheri对确定浓度的6种蛋白混合物的分析表明MALDI-TOF-MS/MS的鉴定结果更可靠,而在分析复杂的生物样品时MALDI-TOF-MS/MS与ESITOF-MS/MS相差不多[8-9]。经液相色谱分离的肽段MALDI-TOF-MS/MS通常比ESI-TOF-MS/MS更常用。传统iTRAQ不能联用含离子阱的质谱仪如LTQ-Orbitrap进行蛋白定量,这是由于在标准的MS/MS碎片离子碰撞诱导解离(CID)下,质谱仪中的离子阱不能识别低分子量阈值的小产物离子。有人利用LTQ-Orbitrap质谱仪,通过改进脉冲解离(PQD)和高能碰撞诱导解离(HCD)片段方法,与CID联合,使iTRAQ全蛋白组定量更敏感更准确[10]。iTRAQ分析结果经生化检测方法如定量Western印迹和免疫荧光显微技术验证有着很高的相关性,但提供合适的iTRAQ数据统计分析也是必要的[11]。无论使用什么质谱仪,低信号数据都有相对较高的变异性[12]。低丰度蛋白常以小肽段形式检测,但当使用基于统计的滤波方法时,小肽段会被数据库漏掉。许多生物模型有助于解决这些问题,包括峰值强度的加乘误差模型和IsobariQ元件采用的方差稳定规范化(VSN)算法[13]。同位素污染和背景干扰常会引起iTRAQ值的一系列低估。利用数据处理软件校正化学浓缩和iTRAQ试剂中天然同位素杂质,从而可提供相对准确的同位素标记。在前体离子选择期间,质谱仪不能识别相似质荷比的2个肽段,这些肽段产生的二级质谱光谱将会包含碎片离子和iTRAQ报告基团,通过高分辨率的样品分离,可以缓解相似肽段之间的覆盖[14]。
5 iTRAQ分析结果的校验
iTRAQ能够无偏见地定量和分析样品蛋白组信息,其目的是为我们进一步研究提供线索,而不是提供最终结论。iTRAQ实验产生大量数据,须提取有用信息并进一步验证,验证方法取决于实验目的,但一些关键点应考虑。用cut-off值去除检测低于确定数目和离子总量分数置信区间的肽段,可大大简化数据分析(检测低于95%总离子分数置信区间和小于2肽)。iTRAQ蛋白率是平均值,通过计算每个肽段的单个肽段率来鉴定蛋白。任何一个不准确的肽段率可能会改变整个蛋白的平均比值,因此须密切关注每个蛋白的单个肽段率。iTRAQ检测的蛋白差异表达情况可通过生化方法进行验证,如Western印迹、ELISA或免疫组化。当免疫学方法不可用时,基于多反应监测(MRM)的高通量质谱能有效用于验证生物标记物[15]。
6 iTRAQ揭示病毒致病机制
基于质谱技术的定量或半定量蛋白组方法,已广泛应用于病毒宿主相互作用的研究中,针对病毒感染的宿主研究阐释了诸多病毒宿主相互作用的分子事件,为揭示病毒致病机制和诊断治疗提供了重要线索。
Lu[16]等用iTRAQ技术比较了猪繁殖与呼吸综合征病毒(PRRSV)感染和非感染条件下,猪肺泡巨噬细胞(PAM)的蛋白表达情况,共鉴定到160个宿主蛋白在病毒感染后明显改变,这些差异表达蛋白参与病毒结合、细胞结构、信号转导、细胞黏附等多个生物过程,他们通过Western印迹对IFIT3、SOD2和hnRNP A1等3个蛋白的表达进行了验证,发现hnRNP A1在PRRSV的宿主细胞内复制中扮演重要角色。Liu[17]等用相同方法比较了猪圆环病毒感染猪肺泡巨噬细胞的蛋白组学变化,共鉴定到145个宿主细胞蛋白发生了差异表达,这些差异表达蛋白对于阐明病毒复制和致病机制具有重要作用。Li[18]等对石斑鱼虹彩病毒感染石斑鱼胚胎细胞系做了iTRAQ分析,鉴定到49个病毒蛋白,其中11个是首次报道。另外还鉴定到743个宿主蛋白,这些蛋白可归类到218个不同的蛋白分类中,其中分别有14个和5个宿主蛋白在病毒感染后出现表达上调和下调。α-2-巨球蛋白同型体1参与蛋白酶抑制,该蛋白在石斑鱼虹彩病毒感染石斑鱼胚胎细胞后表达上调,表明病毒可能通过抑制宿主蛋白酶和泛素表达而保护自身蛋白组分不被降解。iTRAQ技术分析结果为进一步研究和了解病毒宿主的相互作用、病毒感染的分子机制和发病机理提供了重要线索。
iTRAQ技术也可用于全病毒的组分分析。Li[19]等用iTRAQ技术联合LC/MALDI-TOF MS/MS对对虾白斑综合征病毒(WSSV)做了蛋白质组学分析,发现7个蛋白具有乙酰化的N端,RT-PCR确定了13个新鉴定的病毒蛋白。而且利用iTRAQ技术可区分WSSV病毒粒子膜蛋白与核衣壳蛋白,共鉴定到23个包膜蛋白和6个核衣壳蛋白,提示iTRAQ是一种能有效鉴定病毒蛋白亚细胞定位的方法。
7 iTRAQ在医药领域的应用
近年来iTRAQ技术在癌症、神经退行性疾病、肝脏和肾脏问题、妊娠毒血症、糖尿病、胰腺炎和自身免疫性疾病等医疗领域应用广泛。通过研究发现疾病标记物,以便弄清疾病的发病机制,提供疾病早期诊断方法,鉴定潜在的治疗靶点,以及弄清药物的作用机制。还有研究试图找到一些标记物用于预测癌症病人预后[20-21]。
DeSouza等[22]利用iTRAQ试剂定量标记正常子宫和子宫癌组织,鉴定到9个潜在的生物标记物,后试图采用40份临床样品验证这一结果,最后发现用这9个标记物区别正常和癌症病人时并不具备敏感性和特异性,但却发现另外3个蛋白(丙酮酸激酶、伴侣蛋白10和α1-抗胰蛋白酶)有着很好的敏感性和特异性。Richard[23]等用iTRAQ试剂对白血病癌基因TEL/PDGFRβ转染和未转染的干细胞系(FD⁃CP2mix)裂解液进行标记,并用液相色谱进行检测,根据报告离子的比值得到蛋白表达量的变化,并与用微阵列技术检测的转染和对照干细胞系结果进行对比,具有一致性,可能对癌基因作用机制研究有帮助。Glen等[24]利用iTRAQ鉴定到前列腺癌退化抗原gp96,一种对区分恶性和良性肿瘤有重要意义的标记物。Rudrabhatla[25]等利用iTRAQ技术发现神经微丝蛋白氨基酸残基在Alzheimer病人中有着显著的磷酸化。用iTRAQ也可以鉴定到膜蛋白,Han等[26]利用iTRAQ比较了常染色体显性多囊肾病野生型和患病鼠模型的肾脏质膜,鉴定到潜在的治疗靶标。Grant[27]等利用iTRAQ研究心脏左心室衰老后蛋白组变化,揭示随着年龄的增长心脏失去舒张功能的机制。Pendyala[28]等利用iTRAQ研究HIV-1感染后出现的神经认知性障碍(HAND)的蛋白组学变化,发现维生素E结合蛋白Afamin表达下调。
利用iTRAQ比较正常与病理情况下蛋白组学变化,对于诊断疾病具有重要意义。但体内皮肤成纤维细胞、血清、唾液和其他组织常因病人年龄、性别和基因背景不同而出现变化,而不是由于基因突变或疾病本身状态导致的特异性。例如,Miike[29]等利用iTRAQ技术展现了血清蛋白组分的性别差异;Truscott[30]等利用iTRAQ分析人类水晶体,表明随着年龄差异,蛋白-膜的相互作用有显著改变。在关于遗传神经肌肉疾病的研究中,脊髓性肌萎缩(SMA)病人的GM03813皮肤成纤维细胞基因突变导致SMN蛋白大量减少。结合iTRAQ标签技术和二维液相色谱及MALDI TOF/TOF质谱分析,比较SMA病人和正常人的成纤维细胞的定量蛋白质组变化,发现SMA病人和相同年龄的阴性组最大的差异是他们具备不同基因型。但是,存在于惟一原代细胞系的肌源细胞(GM03813)而不是其他因素导致成肌细胞特定蛋白如SMA细胞中的肌间线蛋白显著增加。这些现象为我们获得无成肌细胞的纤维细胞群体的无限增殖和克隆原代细胞系提供了线索[11]。
在体内进行药物对病人影响的研究非常困难,面临诸多问题,如组织样品采集、饮食改变、感染和病人年龄等,但通过药物和非药物作用单细胞系的iTRAQ比较用于研究药物作用和影响机制则直截了当。Wang[31]等利用iTRAQ技术研究β-受体阻滞药卡维地洛对血管平滑肌细胞的影响,发现13个蛋白发生了差异表达。Bai等利用iTRAQ技术观察抗凝结药物华法林对HepG2细胞的影响,鉴定到2种蛋白DJ-1和14-3-3发生了明显的差异表达。2-丙基戊酸钠被用作抗惊厥和心境稳定剂,是一种脱乙酰化酶(HDAC)抑制剂,被认为是治疗SMA的有效药物,常见副作用是导致骨质疏松。为了探清药物治疗SMA出现的副作用情况,利用iTRAQ技术对药物治疗和无药物治疗的SMA皮肤成纤维细胞进行定量蛋白组学研究,结果发现治疗组的胶原Ⅰ和Ⅵ、胶原结合糖蛋白、骨钙化素显著减少。胶原Ⅰ是骨基质的主要蛋白成分,骨粘连蛋白在骨形成中扮演重要角色,这些结果揭示了长期服用2-丙基戊酸钠后导致出现骨质疏松的机制。
8 生物靶标
尽管已有很多文献报道利用iTRAQ鉴定到潜在疾病标记物,但是只有很少的靶标得到充分验证,可以作为临床患者受益的靶标。为了让一个新的生物标记物引入临床实践,必须提供充分的证据来证明该标记物是有效的、精确的和可重复的,并且它除了能够有助于诊断病情或改善病情状况,也应当有成本效益[32]。
9 结语
毋庸置疑,最近iTRAQ标签技术对定量蛋白组学的发展有着重要的影响。在同一实验中可分析8种样品增加了实验设计的灵活性,并且没有复杂的质谱数据分析。新的生物标记的发现将帮助我们更好地弄清疾病致病机制和预后,用于提高早期诊断的敏感性,找到治疗靶点,弄清药物治疗作用的机制。尽管iTRAQ在发现潜在生物标记物中有着重要作用,但将iTRAQ技术用于临床实验室的日常分析仍需要解决众多问题。随着影响iTRAQ定量准确性问题的解决和数据分析工具的改进,在未来几年内医学研究将从iTRAQ定量蛋白质组技术中得到巨大的帮助。
[1] Gygi S P,Rist B,Gerber S A,et al.Quantitative analysis of complex protein mixtures using isotope-coded affinity tags[J].Nat Biotechnol,1999,17(10):994-999.
[2] Wu W W,Wang G,Baek S J,et al.Comparative study of three proteomic quantitative methods,DIGE,cICAT,and iTRAQ,using 2D gel or LC-MALDI TOF/TOF[J].J Proteome Res,2006,5(3):651-658.
[3] Ong S E,Blagoev B,Kratchmarova I,et al.Stable isotope la⁃beling by amino acids in cell culture,SILAC,as a simple and accurate approach to expression proteomics[J].Mol Cell Proteomics,2002,1(5):376-386.
[4] Ross P L,Huang Y N,Marchese J N,et al.Multiplexed pro⁃tein quantitation in Saccharomyces cerevisiae using amine-re⁃active isobaric tagging reagents[J].Mol Cell Proteomics,2004,3(12):1154-1169.
[5] Choe L,D'Ascenzo M,Relkin N R,et al.8-Plex quantita⁃tion of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease[J].Proteomics,2007,7(20):3651-3660.
[6] Fuller H,Morris G.Quantitative proteomics using iTRAQ la⁃beling and mass spectrometry[M]//Leung H E.Integrative pro⁃teomics.Croatia:InTech,2012:347-362.
[7] Washburn M P,Wolters D,Yates J R.Large-scale analysis of the yeast proteome by multidimensional protein identifica⁃tion technology[J].Nat Biotechnol,2001,19(3):242-247.
[8] Kuzyk M A,Ohlund L B,Elliott M H,et al.A comparison of MS/MS-based,stable-isotope-labeled,quantitation perfor⁃mance on ESI-quadrupole TOF and MALDI-TOF/TOF mass spectrometers[J].Proteomics,2009,9(12):3328-3340.
[9] Scheri R C,Lee J,Curtis L R,et al.A comparison of rela⁃tive quantification with isobaric tags on a subset of the mu⁃rine hepatic proteome using electrospray ionization quadrupole time-of-flight and matrix-assisted laser desorption/ionization tandem time-of-flight[J].Rapid Commun Mass Sp,2008,22(20):3137-3146.
[10]Köcher T,Pichler P,Schutzbier M,et al.High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap:a new mass spectrometric method combining the benefits of all[J].J Proteome Res,2009,8(10):4743-4752.
[11]Fuller H R,Man N T,Lam L T,et al.Valproate and bone loss:iTRAQ proteomics show that valproate reduces collagens and osteonectin in SMA cells[J].J Proteome Res,2010,9(8):4228-4233.
[12]Karp N A,Huber W,Sadowski P G,et al.Addressing accura⁃cy and precision issues in iTRAQ quantitation[J].Mol Cell Proteomics 2010,9(9):1885-1897.
[13]Arntzen M Ø,Koehler C J,Barsnes H,et al.IsobariQ:soft⁃ware for isobaric quantitative proteomics using IPTL,iTRAQ,and TMT[J].J Proteome Res,2010,10(2):913-920.
[14]Ow S Y,Salim M,Noirel J,et al.Minimising iTRAQ ratio compression through understanding LC-MS elution depen⁃dence and high-resolution HILIC fractionation[J].Proteomics,2011,11(11):2341-2346.
[15]Anderson L,Hunter C L.Quantitative mass spectrometric mul⁃tiple reaction monitoring assays for major plasma proteins[J].Mol Cell Proteomics,2006,5(4):573-588.
[16]Lu Q,Bai J,Zhang L,et al.Two-dimensional liquid chroma⁃tography-tandem mass spectrometry coupled with isobaric tags forrelative and absolute quantification(iTRAQ)labeling ap⁃proach revealed first proteome profiles of pulmonary alveolar macrophages infected with porcine reproductive and respirato⁃ry syndrome virus[J].J Proteome Res,2012,11(5):2890-2903.
[17]Liu J,Bai J,Lu Q,et al.Two-dimensional liquid chromatog⁃raphy-tandem mass spectrometry coupled with isobaric tags forrelative and absolute quantification(iTRAQ)labeling ap⁃proach revealed first proteome profiles of pulmonary alveolar macrophages infected with porcine circovirus type 2[J].J Pro⁃teomics,2013,79:72-86.
[18]Chen L M,Tran B N,Lin Q,et al.iTRAQ analysis of Singa⁃pore grouper iridovirus infection in a grouper embryonic cell line[J].J Gen Virol,2008,89(Pt 11):2869-2876.
[19]Li Z,Lin Q,Chen J,et al.Shotgun identification of the struc⁃turalproteome ofshrimp white spotsyndrome virus and iTRAQ differentiation of envelope and nucleocapsid subpro⁃teomes[J].Mol Cell Proteomics,2007,6(9):1609-1620.
[20]Matta A,Tripathi S C,DeSouza L V,et al.Heterogeneous ri⁃bonucleoprotein K is a marker of oral leukoplakia and corre⁃lates with poor prognosis of squamous cell carcinoma[J].Int J Cancer,2009,125(6):1398-1406.
[21]Tripathi S C,Matta A,Kaur J,et al.Nuclear S100A7 is asso⁃ciated with poor prognosis in head and neck cancer[J].PloS One,2010,5(8):e11939.
[22]DeSouza L V,Grigull J,Ghanny S,et al.Endometrial carcino⁃ma biomarkerdiscovery and verification using differentially tagged clinical samples with multidimensional liquid chroma⁃tography and tandem mass spectrometry[J].Molecular&Cellu⁃lar Proteomics,2007,6(7):1170-1182.
[23]Unwin R D,Pierce A,Watson R B,et al.Quantitative pro⁃teomic analysis using isobaric protein tags enables rapid com⁃parison of changes in transcript and protein levels in trans⁃formed cells[J].Mol Cell Proteomics,2005,4(7):924-935.
[24]Glen A,Gan C S,Hamdy F C,et al.iTRAQ-facilitated pro⁃teomic analysis of human prostate cancer cells identifies pro⁃teins associated with progression[J].J Proteome Res,2008,7(3):897-907.
[25]Rudrabhatla P,Grant P,Jaffe H,et al.Quantitative phospho⁃proteomic analysis of neuronal intermediate filament proteins(NF-M/H)inAlzheimer'sdiseasebyiTRAQ[J].FASEB J,2010,24(11):4396-4407.
[26]Han C L,Chien C W,Chen W C,et al.A Multiplexed quan⁃titative strategy for membrane proteomics opportunities for min⁃ing therapeutic targets for autosomal dominant polycystic kid⁃ney disease[J].Mol Cell Proteomics,2008,7(10):1983-1997.
[27]Grant J E,Bradshaw A D,Schwacke J H,et al.Quantifica⁃tion of protein expression changes in the aging left ventricle of Rattus norvegicus[J].J Proteome Res,2009,8(9):4252-4263.
[28]Pendyala G,Trauger S A,Siuzdak G,et al.Quantitative plas⁃ma proteomic profiling identifies the vitamin E binding pro⁃tein afamin as a potential pathogenic factor in SIV induced CNS disease[J].J Proteome Res,2009,9(1):352-358.
[29]Miike K,Aoki M,Yamashita R,et al.Proteome profiling re⁃veals gender differences in the composition of human serum[J].Proteomics,2010,10(14):2678-2691.
[30]Truscott R J,Comte-Walters S,Ablonczy Z,et al.Tight bind⁃ing of proteins to membranes from older human cells[J].Age(Dordr),2011,33(4):543-554.
[31]Wang M,Wang X,Ching C B,et al.Proteomic profiling of cellular responses to Carvedilol enantiomers in vascular smooth muscle cells by iTRAQ-coupled 2-D LC-MS/MS[J].J Proteomics,2010,73(8):1601-1611.
[32]Sturgeon C,Hill R,Hortin G L,et al.Taking a new biomark⁃er into routine use-a perspective from the routine clinical bio⁃chemistry laboratory[J].Proteomics Clin Appl,2010,4(12):892-903.
