酵母表面展示T载体构建及目的蛋白的表面展示
2014-04-29梁秀怡梁志成朱筱刘诗雨张智田生礼
梁秀怡 梁志成 朱 筱 刘诗雨 张 智 田生礼
摘 要:构建一种能对PCR产物进行直接克隆并展示于酵母表面的新型T载体。根据酵母表面展示载体pYD1多克隆位点序列设计出利用两端带有Xcm I内切酶酶切位点的含有黄色荧光蛋白基因的Xcm I酶切盒,通过Nhe I和Xho I酶切位点插入到pYDl裁体上形成质粒pYD-YFP,并对其进行酶切鉴定和DNA测序分析,再经Xcm l酶切后形成两端带有dT的表面展示T载体。利用PCR扩增两个含有荧光蛋白的融合蛋白PCAD-CFP和PSR-DsRed的基因并直接克隆到所构建的T载体中,检测其表达功能。 酶切鉴定和DNA测序结果显示PCAD-CFP和PSR-DsRed正确插入载体上,分别转化至酿酒酵母EBY100中,激光共聚焦显微镜下观察到相应的荧光的酵母,表明克隆有融合蛋白基因片段的载体成功在酵母细胞中进行表面展示,证明了所构建的酵母表面展示T载体具有直接克隆和表面展示目的蛋白的功能。
关键词:T栽体:酵母表面展示;真核表达:Xcm I酶切盒
中图分类号:Q782
文献标识码:A
文章编号:1007-7847(2014)()6-()477-06
酵母表达系统具有培养成本低廉、便于大规模培养和培养周期短等特性,同时又具有真核生物的蛋白翻译后修饰功能,近年来逐渐用于药用和食用蛋白的生产。酵母表面展示技术(yeastsurface display system)在20世纪90年代初开始兴起,是-种将外源蛋白质进行固定化表达的真核展示系统.广泛应用于蛋白质的相互作用、抗体研制、蛋白分子定向等领域,特别在生物乙醇生产过程中起到关键作用,包括淀粉酶、纤维素酶等酶蛋白均已进行表面展示。 a凝集素和α凝集素是锚定在细胞壁上的甘露糖蛋白,介导单倍体交配型(MATa)和(M ATα)的识别和粘附。最常见酵母表达系统为a凝集素展示系统和α凝集素展示系统,a凝集素由两个亚单位Agalp和Aga2p组成,由于Agalp亚基通过与细胞壁上的β葡聚糖的共价连接而锚定在细胞壁,Aga2p和Agalp两个亚基通过两对二硫键相连,因此a展示系统是将a凝集素的Aga2p亚基与目的蛋白进行融合,在信号肽的引导下实现酵母的表面展示。α凝集素展示系统则将目的蛋白与α凝集素融合,从而将其展示于酵母细胞表而。其中酵母表而展示主要采用的宿主菌是酿酒酵母(Sacclzaromyces cerevisiae),已被FDA列为CRAS (Generally regarded as safe)原材料.酿酒酵母表面展示能够把日的蛋白与凝集素功能结构域融合,将目的蛋白表达并定位于酵母细胞膜的表面,不但易于纯化鉴定,并且酵母具有真核生物的翻译后修饰体系,能表达具有一定活性的真核生物的蛋白。相比细胞内表达的酶.酵母表面展示可多次生产展示于细胞表面的蛋白,并且在廉价的培养基中培养可达到很高的细胞密度,适应工业生产需求,省去酶的纯化和固定化等繁琐步骤。
构建酵母表而展示载体是蛋白质进行酵母表面展示的基础,PCR产物克隆需耗费大量的时间与精力在表达载体的构建、质粒的提取和重组子的鉴定上,而利用T载体可对PCR产物进行快速有效的克隆,因此有必要构建一个可直接对PCR产物进行克隆和表达的酵母表面展示T载体,特别是进行大量的PCR产物克隆时,可以极大减少实验操作的步骤。
本文克服了常规构建表达载体操作繁琐的缺点,以酵母表面展示载体pYDl为基础,通过向载体pYDl插入含有黄色荧光蛋白基因的Xcm I酶切盒,再切除黄色荧光蛋白基因来构建酵母表面展示T载体,并利用荧光蛋白基因检测该表面展示载体的克隆和表达功能。
1 材料与方法
1.1材料
1.l.l 菌株和载体
克隆载体pMD-18T购自日本Takara公司;pDsRedl-Nl、pEYFP-Cl、pECFP-Cl、pYD-l载体购自美国Invitrogen公司,大肠杆菌JM107和酿酒酵母EBY100由本实验室保存。
1.1.2主要试剂
Taq DNA聚合酶、T4 DNA连接酶购于日本rllakara公司;限制性核酸内切酶Nhe I、Nde I、Xho I、BamH I、Xcm I为美国NEB(北京)公司产品;所有PCR反应的引物(表1)及测序均在北京华大基因公司合成。
1.2方法
1.2.1 Xcm I酶切盒的合成
以含有黄色荧光蛋白基因的质粒pEYFP-Cl为模板,使用的引物pYDTl和pYDT2(表1),进行PCR扩增,扩增含有黄色荧光蛋白基因位点的Xcm I酶切盒,长度约760 bp,扩增程序为:预变性94℃ 5 min; 94℃ 30 s,55℃ 50 s,72℃ 10 min,进行30个循环,72 ℃延伸7 min。PCR产物进行l%琼脂糖凝胶电泳后切胶回收后,通过T4 DNA连接酶连接至pMD-18T裁体,重组载体命名为pMD-YFP,并送往华大基因公司进行序列测定,分析克隆基因序列组成及其阅读框架的正确性。
1.2.2 酵母表面展示T载体pYD-T的构建及鉴定
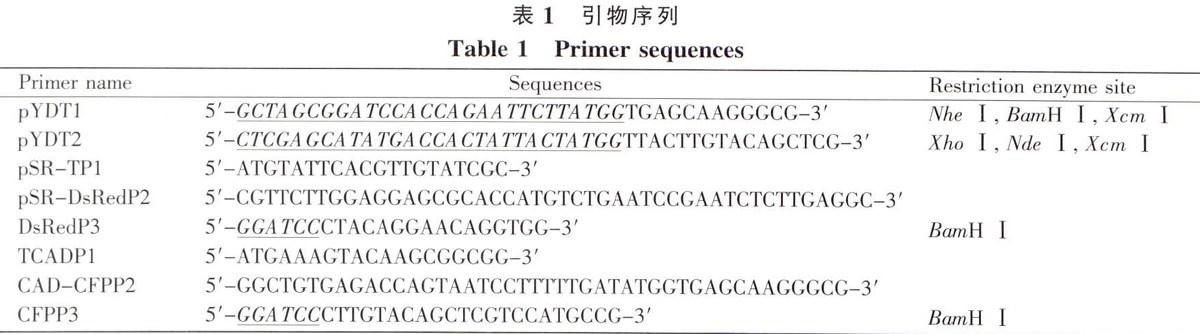
用限制性内切酶Xho 1和Nhe l酶切pMD-YFP后进行电泳,切胶回收切出的小片段,连接到经由同样的限制性内切酶酶切pYDl质粒上,获得的重组质粒命名为pYD-YFP,质粒及多克降位点序列图谱见图l。将pYD_YFP转入大肠杆菌JM107感受态细胞中,涂布于含50 μg/mL氨苄青霉素抗性的LB平板,37℃培养过夜后挑取单菌落提取质粒.对酶切鉴定正确的质粒再进行DNA测序分析将DNA测序正确的pYD-YFP转化至酵母EBY100感受态细胞,铺于SD/Trp-平板上培养,挑取直径为2~3 mm的酵母EBYIOO单菌落于于10 mL,酵母SD/Trp'液体培养基(含2%乳糖)中进行诱导表达.30℃,200 r/min条件下培养48~72 h。取诱导后的酵母菌液300μL,1 000 r/min离心5 min,去上清液,用lmL无菌水洗涤并重悬菌体,由于原载体pYD-PYDl是酿酒酵母表而展示载体、改造后的载体上插入带有YFP基因的酶切盒片段,YFP蛋门通过锚定蛋白展示到酵母细胞壁上,因此在505 nm激发光下通过激光共聚焦显微镜可直接观察到相应的荧光,证明了载体pYD-YFP的正确性。质粒pYD-YFP经Xcm I酶切后,回收载体部获得酵母表而展示T载体。
1.2.3 酵母表面展示T载体pYD-T克隆目的基因
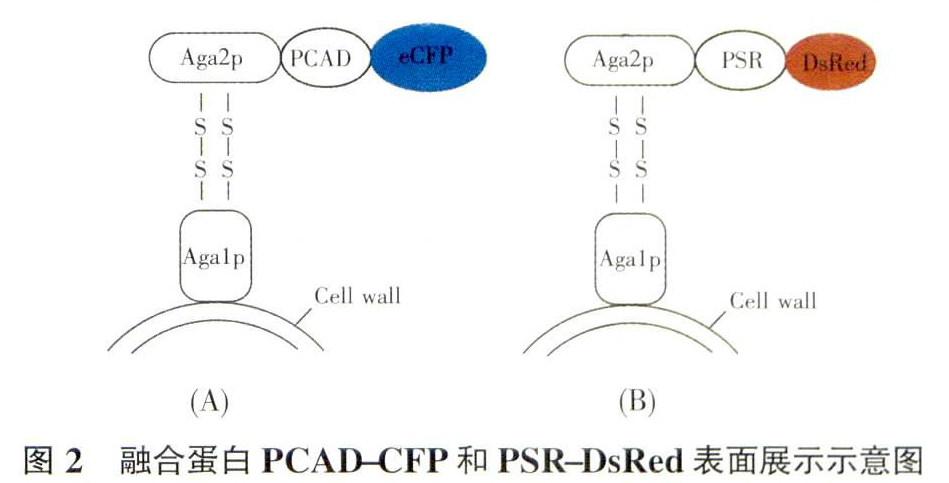
本文利用多头绒泡菌(Physarum, poly-cephalun)的两个蛋白肉桂醇脱氢酶PCAD (Gen-Bank登陆号:KF861979.1)和PSR蛋白(GenBank登陆号:FJ917746)对pYD-T的功能进行验证。使用引物TCADP1、CAD-CFPP2和CFPP3通过重叠PCR将PCAD与青色荧光蛋白eCFP的两个基因连接构成融合基冈PCAD- CFP。另外使用引物pSR -TPl .pSR -DsRedP2和DsRedP3通过重叠PCR将PSR和红色荧光蛋门DsRed两个基因连接构成融合基因PSR-DsRed,以上两个融合基因基因片段3端均引入一个BamH I酶切位点。
将pYD-T载体用限制性内切酶Xcm l酶切处理,经琼脂糖凝胶电泳后回收载体部分,得到两端带有突同T的线性化T载体,分别连接PCAD-CFP和P.SR-D.s Red,获得的重绀载体分别命名为pYD-PSR-DsRed和pY D—PCAD一CFP,融合蛋白表面展示示意图见图2。将连接好的重组载体转入大肠杆菌JM107感受态细胞中,涂板37℃培养过夜。挑取长势良好的单菌落接种于LB液体培养基中培养,提取质粒DNA进行酶节鉴定。
1.2.4 酵母表面展示T载体pYD-T表达功能验证
将pYD -PSR-DsRed和pYD - PCAD一CFP重组载体分别转化至酿酒酵母感受态细胞中,操作过程参见文献,将菌液铺于SD/Trp-培养板,30℃培养3 d,通过菌液PCR筛选阳性转化子。将阳性转化子接种于酵母SD/Trp-液体培养基(含2%乳糖)中,30 ℃, 200 r/min培养48—72 h取适量的菌液在激光共聚焦显微镜下观察拍照。
2 结果与分析
2.1 酵母表面展示T载体pYD-T载体的构建
将Xcm I酶切盒片段克隆到pMD-18T载体中形成重组载体pMD-YFP.用限制性内切酶Nhe I和Xho I酶切鉴定,结果见图3泳道1、2、3,酶切出的片段与Xcrn I酶切盒长度相近,约为760 bp。Xcm I酶切盒片段通过Nhe I和Xha I 位点插入酵母表而展示表达载体pYD-l中,得到载体pYD-YFP重组,酶切鉴定结果见图3泳道4 ,5;图3泳道6为重组载体pMD-YFP经过Xcm I酶切出YFP皋因后,回收载体部分获得酵母表面展示T载体pYD-T。DNA测序结果表明所构建的真核表达载体含有完整的黄色荧光蛋白位点及酶切盒,证明pYD-YFP载体构建成功。pYD-YFP质粒转化至酿酒酵母EBY 100后经SD诱导培养基诱导72 h后.在激光共聚焦显微镜下拍摄到的细胞表面展示的YFP蛋白,图4中均能清晰看到酵母表面展示呈现为绿光,这是因为带有黄色荧光蛋白基因的酵母在青光的激发下呈现两种光的合成光.即为绿色,部分酵母表面的绿色呈光圈状,这说明Xcm I酶切盒中黄色荧光蛋白YFP已经在酵母细胞膜上展示。
2.2 酵母表面展示T载体pYD-T克隆功能的鉴定
目标基因片段用PCR扩增方法在5或3端引入一个与T载体的3或5端所对应相同的限制性内切酶位点(该限制性内切酶位点务必确保在PCR片段中或者在T载体有且仅有一个),本文在PCAD-CFP和PSR-DsRed基因片段3端引入一个BamH I酶切位点,与载体5端的BamH I酶切位点相对应.连接目的基因的PCR产物与T载体构成重组载体,利用所设的限制性内切酶进行单酶切鉴定。本研究使用BamH I进行单酶切,如果能切出与目标基因长度大小相近的片段,即可鉴定为正向连接,若没有切出相应大小的DNA片段,则是反向连接,因为实际上反向连接时应该切出约为20 bp左右的片段,由于片段太小电泳后兀法直接观察到,图5、6显示BamH I酶切后得到与重叠PCR产物大小相近的条带,表明CAD-CFP和PSR-DsRed均载体pYD-T正向连接,DNA测序结果同样证明了重组载体pYD-CAD-CFP和pYD-PSR-DsRed构建正确。
2.3 酵母表面展示T载体pYD-T的展示功能验证
将重组载体pYD-CAD-CFP和pYD- PSR -DsRed转化至酿酒酵母BY100感受态细胞中,挑取单菌落后接种至在SD/Trp-液体培养基(含20乳糖)中,30 ℃中震荡培养3d后在激光共聚焦显微镜下的观察展示结果,分别用558nm和405nm激发光检测,可以看到酵母的表面发出明亮的红光、青光,见图7、8结果显示目标蛋白主要分布在酵母细胞的表面,形成明显的青色或红色光圈,证明CAD-CFP、PSR-DsRed融合蛋白已经成功在酵母表而展示。
3 讨论
近10年来,酵母表面工程技术在生物技术领域得到迅速发展,酵母表面展示系统不仅能展示单个亚单位蛋白,而且能展示异源寡聚体多亚单位蛋白,在展示高等哺乳动物蛋白天然构象方而具有其独特的优越性。 T载体广泛应用于PCR产物克隆,目前商品化的T载体主要是克降型T载体,只能用于基因的克隆与保存,不能用于基因的表达。若要进行目的基因的表达,需另外将目的基因克隆至表达载体,过程中涉及转化子的大量测序和鉴定操作。本研究利用可变内切酶Xcm I,识别的序列为CCA (N5/N4) TGG,将识别序列第8位没为T,酶切盒上包含两个反向的Xcm l酶切位点,经Xcm I酶切后可产生3'带有一个突出dT的T载体。两个Xcm I位点之问包含黄色荧光蛋白基因,目的在于通过转化测定菌体的荧光来检测Xcm I的酶切效率,从而提高所制备T载体的质量。PCR产物可以无需酶切、纯化,减少筛选有效重组子的过程,通过TA克隆,直接连接到pYD-T载体构成重组载体,无需另外将目的基因重组至表达载体,直接进行蛋白在酿酒酵母表面展示(图7、8),节省宝贵的研究时间和经费,通过克降和表达功能的鉴定后证明此方法新颖可行:
目的基因在构建表达载体时,一般通过双酶切法将目的基因克隆到表达载体,因此需要考虑目的基因上是否含有载体多克隆位点的酶切识别位点。通过酶切位点插入到载体上的过程中应避免使用目的基因上已有的酶切识别位点。本文在研究融合蛋白PCAD-CFP和PSR-DsRed在酵母表面的展示过程中,发现载体上的多克隆位点均出现目的基因上,难以通过传统双酶切法克隆至表面展示载体pYDl上,文中通过对pYDl载体进行改造后制备出T载体,除了内切酶Xcm I外,无需考虑载体与目的基因上的其他酶切位点,重组的过程不需要考虑使用何种相应的限制性内切酶,极大降低目的基因序列中含有与载体多克隆位点所用限制性内切酶冲突的可能。通过TA可以连接,成功将目的基因克隆到载体pYD-T上,一步构建表达载体。若使用高保真Taq酶扩增,PCR产物不具有dA粘性末端,可再使用Taq酶进行扩增,使得PCR产物3'端带有dA,这样既可减少PCR过程中的错配率,也可提高PCR产物与载体的连接效率。
本文所构建的酵母表面展示T载体具有操作简便.快捷高效的特点,使具有药用和工业应用价值的蛋白能够更简捷地进行酵母表面展示,为促进更多蛋白在酵母细胞表面的展示和酶的固定化研究建立了表达平台。
