离子色谱-串联质谱测定地表水中高氯酸盐
2014-04-26谢永洪
钱 蜀,谢永洪,杨 坪,熊 杰,冉 丹,姚 欢
1.四川省环境监测中心站,四川成都610041
2.四川大学建筑与环境学院,四川成都610065
高氯酸盐是一种强氧化剂,自然界中一般不会自然生成,几乎均为人工合成[1]。由于其强氧化性,高氯酸盐被广泛用于烟花爆竹、火箭燃料、军火工业、安全气囊等领域的爆炸物中[2]。高氯酸盐还具有强稳定性、易溶于水、密度大于水等特点[3],一旦进入水体会持续迁移扩散,污染大范围的地表水源和地下水,且短期内难以降解。由于高氯酸盐本身是一种甲状腺毒素,它会干扰碘的吸收,影响胎儿、儿童脑部的发育,甚至造成脑部的损伤[4-6]。有研究表明,高氯酸盐的半致死量为 404 mg/L[7],7 ~500 μg/kg·d-1不同剂量人类临床实验表明,最低剂量时有1.8%的参与人员甲状腺碘吸收被干扰,而最高剂量则达67.1%[8]。由于监测技术的限制,地表水中的高氯酸盐并没有引起人们的关注,直到1997年美国加州卫生部开发了检出限低至4 μg/L的检测方法,各地环境水体中才陆续检验出了高氯酸盐的存在。如加州供水公司发现在110个水井中共计有33个水井中含有浓度大于18 μg/L的高氯酸根[9];韩国饮用水中的高氯酸盐质量浓度为0.15 ~35.0 μg/L[10];印度 6 个州 13 个采样点的66个水样高氯酸盐含量范围在未检出~6.9 μg/L之间[11];Wu等[12]在中国15 个城市采集的300 个水样中,有86%的水样都检测出了高氯酸盐。
由于高氯酸盐的强稳定性、易扩散性、毒害性和在环境水体中的普遍检出,各国科研工作者开发了各类方法用于高氯酸盐的分析,美国各州还对饮用水中高氯酸盐允许的最高含量作出了限制。目前,中国还没有相应的标准方法来检测高氯酸盐,亟需建立一套科学、可靠的方法检测水体中的高氯酸盐,为环境执法提供数据支撑和依据。
由于高氯酸盐极易溶于水,溶解后呈稳定的离子状态,因此分析高氯酸盐的方法主要为离子色谱电导检测法。由于高氯酸盐在阴离子交换树脂上的强保留性,不经富集的离子色谱电导检测方法的检出限很低,仅0.1 μg/L左右(25 μL进样体积)[13],必须采用大体积进样(一般为1 mL)才能满足对μg/L级样品的分析,如《离子色谱检测饮用水中高氯酸盐》(US EPA Method314.0),尽管采用了大体积进样,方法的检出限也仅0.5 μg/L,且由于进样体积过大,进入检测体系的大量阴离子在分离柱上过饱和,造成严重拖尾,影响高氯酸盐分析[14],需要采用一定的前处理方法去除。而叶龙等[15]则采用固相萃取方法浓缩样品和去除一定基质以提高离子色谱分析的灵敏度,使50 μL 进样时方法的检出限达 0.15 μg/L。《在线柱浓缩-基体消除离子色谱电导检测饮用水中高氯酸盐》(US EPA Method 314.1)也利用在线浓缩的方法检测高氯酸盐,但分析一个样品的时间很长,达40多分钟。近年来,US EPA开发了二维离子色谱[16]和质谱[17,18]的方法,前者分析时间也需40多分钟,而采用质谱分析则在保证灵敏度的基础上大大节省了分析时间。
在US EPA相应方法的基础上进行了优化和实践,以高容量、强亲水性的IonPac AS16(2 mm)为分离柱,45 mmol/L氢氧化钾为淋洗液,0.3 mL/min的流速,电喷雾负离子模式选择离子监测(SIM),建立了离子色谱-串联质谱(IC-MS)检测高氯酸盐的方法,并用以测定地表水中高氯酸盐,摸清四川省主要河流高氯酸盐含量状况,寻找可能的污染源,控制地表水高氯酸盐污染。
1 实验部分
1.1 仪器与试剂
Dionex ICS-3000离子色谱仪串联MSQ四极杆质谱仪(美国),带有外接 AXP-MS泵,用以泵入有机溶剂,清洗离子源。离子色谱仪带有双泵、双六通阀以同时实现离子色谱电导检测和阀切换切出检测液中弱保留阴离子功能;色谱数据采集和处理采用Chromeleon和Xcalibur色谱工作站;美国Millipore(simplicity 185)超纯水机。
乙腈(HPLC级),高氯酸盐标准溶液(1 000 mg/L,NSI solution公司),超纯水(电阻率为18.2MΩ·cm)。
1.2 实验方法
1.2.1 色谱条件
分析柱为IonPac AS16离子交换柱(2 mm×250 mm),保护柱为 IonPac AG16离子交换柱(2 mm×50 mm),淋洗液为45 mmol/L氢氧化钾,淋洗液流速为0.3 mL/min;柱后辅助试剂为乙腈/超纯水 =1∶1,柱后泵流速为0.3 mL/min,柱温为30℃,检测池温度为35℃,进样体积为68 μL,抑制电导检测(外加水自再生抑制模式,ASRSTM300 2 mm),阀切换时间为8 min,数据采集时间16 min。
1.2.2 质谱条件
-ESI,SIM检测,m/z99.0为定量离子,喷雾温度450℃,锥孔电压70 V,喷雾电压-3 000 V,雾化气5.17×105Pa;质谱采集时间10~16 min。
1.2.3 样品预处理
水样过0.22 μm针式过滤器后直接进样。由于尼龙过滤器对高氯酸盐有一定的吸附作用,过滤水样时采用聚醚砜针式过滤器。
1.2.4 分析流路
超纯水(E1)经泵1(P1)泵入淋洗液自动发生器(EGC)产生分析用的45 mmol/L淋洗液,将由六通阀(V1)进入离子色谱分析系统的样品依次推入保护柱(C0)和(C1),经抑制器(RS)后去除样品中大量阳离子和淋洗液中OH-后进入电导检测器(CD)进行检测,再流入柱后六通阀(V2),通过阀切换使其在0~8 min流入废液,8~16 min流入检测流路,与柱后辅助试剂(乙腈/水=1∶1)混合后进入质谱检测器。同时AXPMS泵(P3)以0.2 mL/min的流速泵入清洗试剂(乙腈/水=1∶1),不断清洗离子源。
分析时,样品进入质谱前先经抑制器后可以去除大量阳离子,再通过六通阀的阀切换功能将其中相对于高氯酸盐而言弱保留的大量阴离子(如氟化物、氯化物、硫酸盐等)切入废液后,在高氯酸盐出峰前再将其切入质谱分析,可以消除离子色谱电导检测时大量阴离子的干扰,同时避免了样品中大量阴阳离子对离子源的污染。分析流程图见图1。

图1 高氯酸盐IC-MS分析流程图
2 结果与讨论
2.1 定量离子的选择
将约1 mg/L高氯酸盐标准溶液注入质谱定量环,经柱后泵入的试剂(乙腈/水=1∶1)推入质谱,负离子模式下进行全扫,得到的质谱图如图2所示,高氯酸盐特征质荷比为98.89(35ClO4-)和100.85(37ClO4-),分析时以灵敏度更高的m/z=99定量,m/z=101定性。
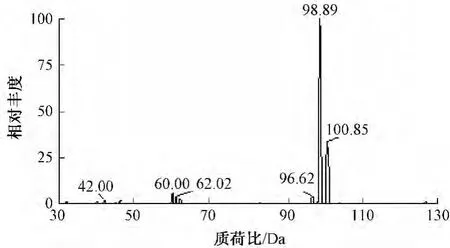
图2 高氯酸盐标准溶液Q1全扫图
2.2 色谱条件的确定
由于高氯酸盐在离子交换树脂柱上的强保留性,选择亲水性强的AS16柱为分析柱,又由于高流速淋洗液不利于质谱分析,因此采用需要较低流速的2 mm内径的AS16柱分析。一般而言,淋洗液流速会极大地影响分析的保留时间,同时也会影响峰形,当流速越高,保留时间越短,峰会更尖锐,但同时峰面积会略为减小。实验时在AS16(2 mm)分析柱能承受的流速范围内讨论了流速对高氯酸盐分析的影响(表1),综合考虑保留时间、峰形和峰面积的影响,选择淋洗液流速为0.3 mL/min。
通常,淋洗液浓度对分析的影响主要体现在对保留时间和峰宽的影响上,对灵敏度影响不大。实验考察了25~65 mmol/L的淋洗液浓度对高氯酸盐离子色谱-质谱分析的影响(表2),综合考虑分析时间与峰宽的影响,淋洗液浓度选择为45 mmol/L。
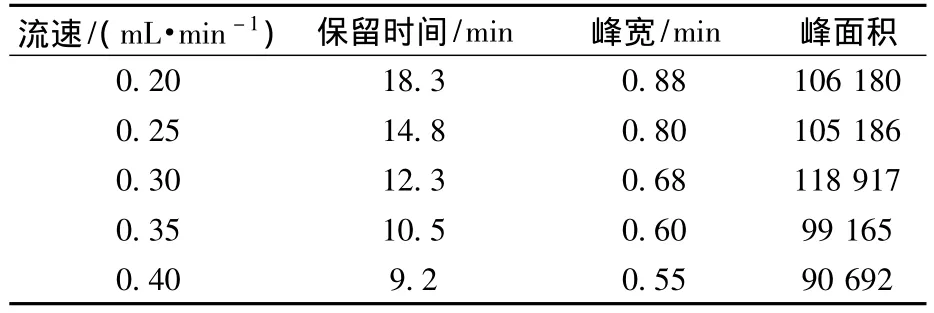
表1 流速对高氯酸盐分析的影响
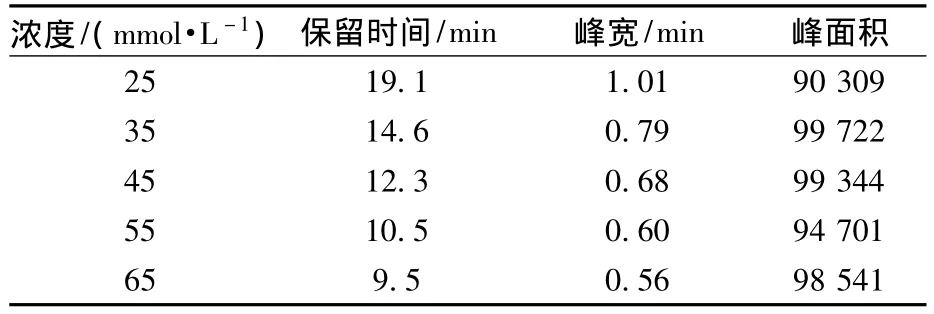
表2 淋洗液浓度对高氯酸盐分析的影响
一般情况下,进样体积越大,方法能检测的样品浓度越低,但进样体积太大,在分析柱柱容量有限的情况下往往会导致柱过饱和,不利于分析。由于采用该法的2 mm内径分析柱柱容量的限制,综合考虑检测要求,进样体积定为68 μL。根据经验,柱温、检测池温度分别设置为30、35℃。
2.3 柱后辅助试剂的使用
IC分离后出来的液体均为无机水相,而由于纯水进入质谱会影响雾化效果,从而影响分析的灵敏度。该法在IC的柱后采用了一辅助试剂泵用于输送乙腈与超纯水的混合液,使之与IC出来的分析液混合后再进入质谱,以增强雾化效果,增加分析的灵敏度。实验时发现,不同的柱后泵流速对分析有一定的影响,但影响不大(图3)。一般而言,在其他分析条件不变的情况下,分析的灵敏度会随着柱后泵流速的增加而增大,但流速过大不适合ESI脱溶剂,抑制库伦爆炸,降低灵敏度,综合考虑仪器耐受压力和灵敏度的问题,柱后泵的流速选择0.3 mL/min。
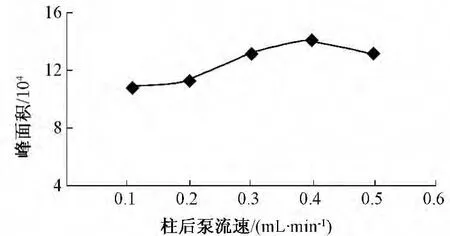
图3 柱后泵流速对高氯酸盐分析的影响
2.4 喷雾温度和锥孔电压的优化
离子源喷雾温度会影响喷雾效果,从而影响分析的灵敏度。实验时在300~550℃范围内考察了温度对高氯酸盐分析的影响。图4显示,当温度从300℃增加到450℃时,峰面积急剧增大,而从450℃增至550℃,峰面积变化不大,因此喷雾温度选择为450℃。
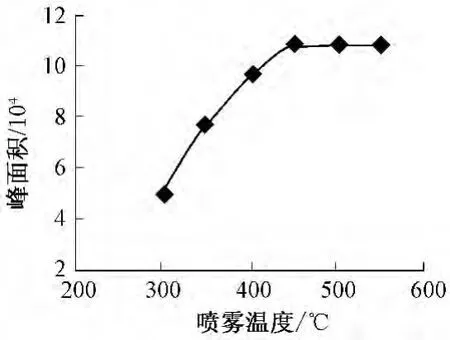
图4 喷雾温度对高氯酸盐分析的影响
锥孔电压也会影响分析的灵敏度,一般锥孔电压选择范围在45~75 V之间,实验结果见图5。
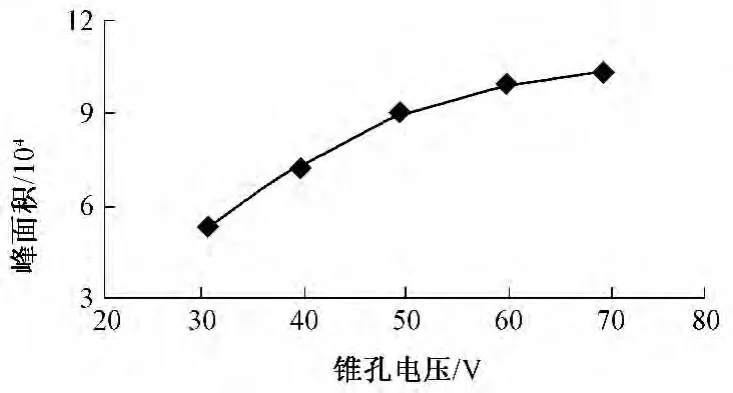
图5 锥孔电压对高氯酸盐分析的影响
当锥孔电压由30 V增加到70 V时,峰面积逐渐增大,但随着锥孔电压的增大,峰面积变化越来越平缓,因此选择70 V作为高氯酸盐分析锥孔电压。
2.5 阀切换时间与质谱采集时间的确定
为了去除水样中大量常见的阴离子(如F-、Cl-、SO42-、NO3-、CO32-、PO43-等),应在样品进入质谱前先通过六通阀切出大量阴离子后再在高氯酸盐出峰前切入质谱分析。因此,实验时均先单独进行离子色谱电导检测分析,根据电导检测的谱图,确定样品切入质谱时间,由于阀切换后样品进入质谱还需一小段时间,质谱采集设置时间应略小于阀切换时间。在上述已经确定的条件下,阀切换的时间设为8 min,质谱开始采集时间设置为10 min。
2.6 方法的线性范围、检出限和精密度
用超纯水配制一系列标准溶液,在上述确定的条件下进样,则该方法在0.1~40 μg/L呈一次曲线,回归方程为y=-407+63 750x,线性相关系数为0.999 9;在0.1~200 μg/L呈二次曲线,回归方程为y=1 577.4+3 454.6x-4.607 1x2,线性相关系数为0.999 7。标准曲线如图6所示。以适当浓度(0.1 μg/L)标准溶液连续进样7次计算标准偏差,乘以3.143[19]得到方法的检出限为 0.031 μg/L。
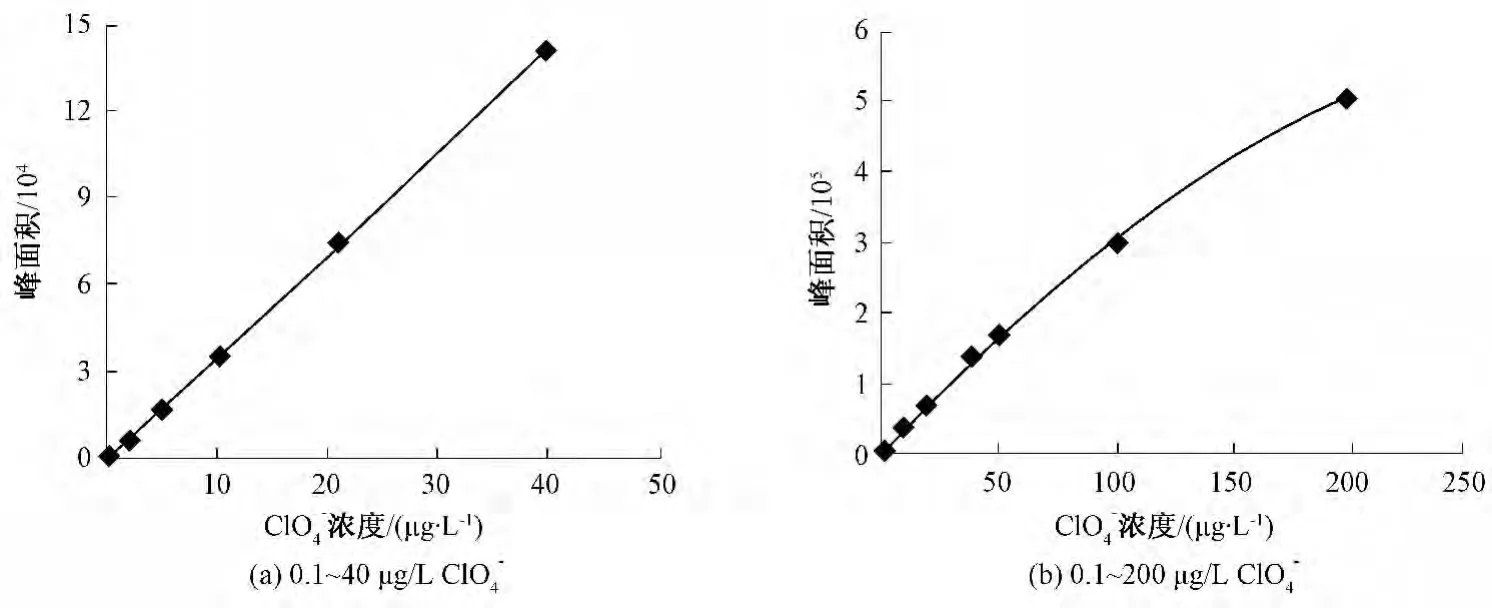
图6 ClO4-标准曲线
在低浓度样品中加入一定量的标准溶液与低、中浓度水平的地表水样品连续进样进行方法低、中、高浓度的精密度实验,结果见表3。低、中、高浓度的地表水样品连续进样6次得到的相对标准偏差为2.26% ~4.45%;分别加入一定量标准溶液后连续进样6次得到平均加标回收率为93.0%~98.0%。可见,对于不同浓度梯度样品,方法的精密度和准确度均较好。
2.7 样品保存时间探讨
将含有不同高氯酸盐的地表水样品经4℃低温冷藏不同天数后,过0.22 μm的聚醚砜针式过滤器,在上述已经确定的条件下进样分析,以考察不同浓度样品保存时间。结果(表4)显示,浓度分别约为1、12、45、93 μg/L的样品保存36 d内 测得的浓度值变化不大。
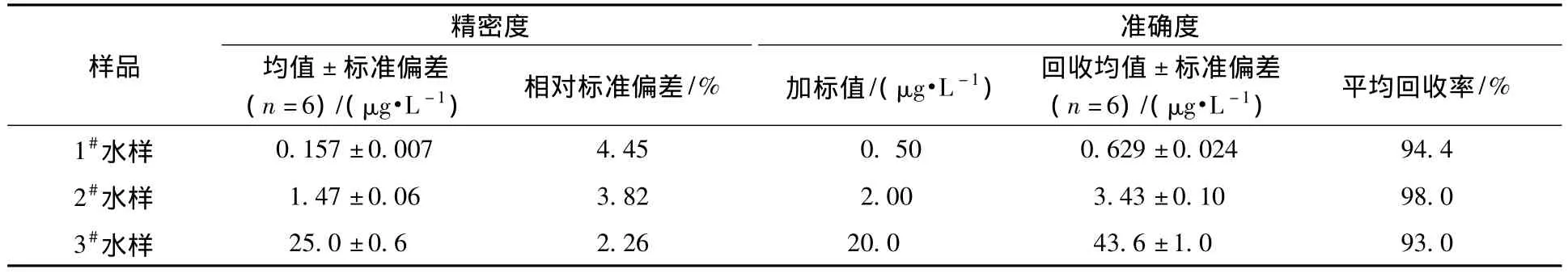
表3 精密度和准确度实验结果
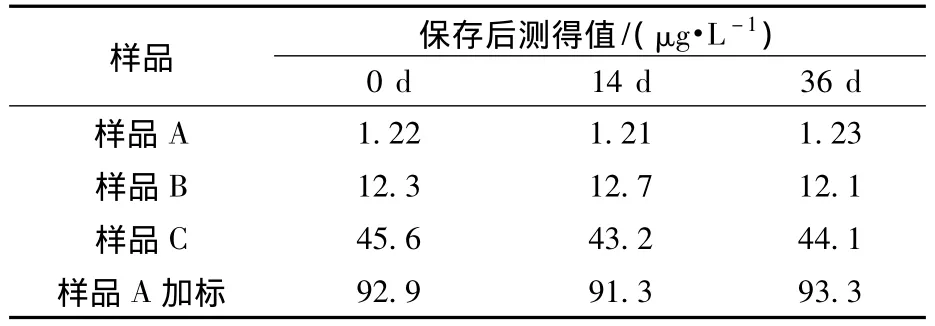
表4 样品保存时间对分析的影响
2.8 比对实验结果
分别用该法与《离子色谱检测饮用水中高氯酸盐》(US EPA Method314.0)的方法(检出限:0.53 μg/L)测定低、中、高不同浓度梯度样品以进行2个方法比对。结果显示,两者分析结果基本吻合,见表5。

表5 比对实验结果 μg/L
2.9 样品分析及来源浅析
2.9.1 样品分析
将2012年7月采集自四川省内主要河流的样品过0.22 μm聚醚砜针式过滤器后直接进样,实验结果见表6。样品浓度范围在未检出(检出限:0.031 μg/L)~ 68.0 μg/L 之间,监测的河流中均含有不同浓度水平的高氯酸盐,其中:河流Ⅱ含量 <0.210 μg/L;河流Ⅰ含量也较低,浓度为0.900~1.30 μg/L;河流Ⅲ和Ⅳ含量较高,浓度为 0.124~68.0 μg/L,除位于上游的一个断面含量(0.124 μg/L)较低外,其余含量均大于10.0 μg/L;河流Ⅴ中也含有一定量的高氯酸盐,从其上游断面至下游的出境断面高氯酸盐含量由3.56 μg/L逐渐降低至0.982 μg/L。
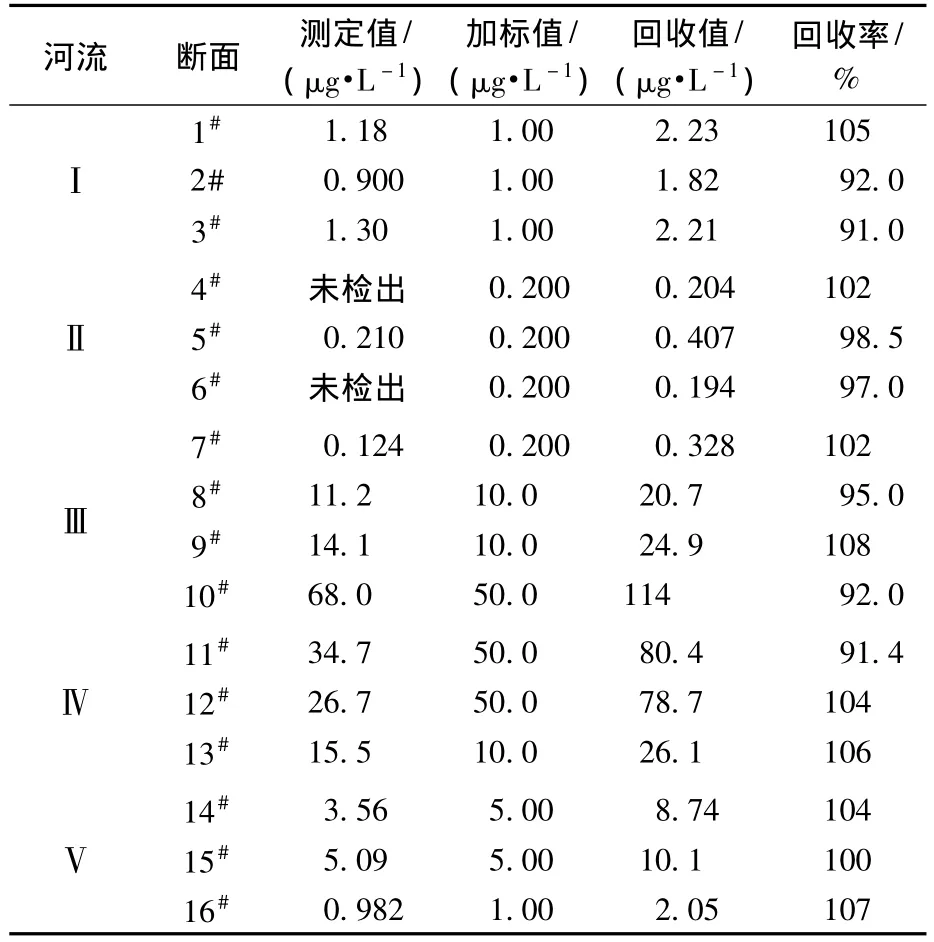
表6 样品分析结果
2.9.2 来源浅析
近年来,各地环境水体普遍受到的污染,美国各州[9,20]、日本[21]、韩国[10]、中国[12]、印度[11]等地均检出不同程度的高氯酸盐,高氯酸盐污染水平从几每升微克至几十每升毫克不等,这些地方水体污染主要来自工业生产。如日本利根川的一支流高氯酸盐含量高达2.3 mg/L,即是受其上游氯酸盐和高氯酸盐生产企业废水排放和推测副产物中含有高氯酸盐的电子企业废水排放影响[21];在美国加利福尼亚州96个水网中的395口井含有高氯酸盐,1997年美国加利福尼亚州洛杉矶县一井水高氯酸盐检出浓度达159 μg/L,被发现附近有Aerojet工厂(生产各种弹药及导弹弹头等)、喷射推进实验室等污染源[20]。
除工业污染源外,高氯酸盐污染还与烟花爆竹的燃放有关。如Kosaka K[21]在同一河流断面刚燃放烟花爆竹时采集一水样,并在5 d后采集一水样,两者浓度含量分别为79、0.39 μg/L。引言中提到的WU等[22]在中国15个城市采集的水样中发现衡阳市高氯酸盐污染较为严重,其饮用水、地下水、地表水中的高氯酸盐质量浓度分别为31.4 ±21.3、22.1、51.3 ±4.4 μg/L,原因是当地有很多大型的烟花爆竹生产加工厂。也有研究[22]显示,烟花爆竹燃放后的城市空气中PM10~PM100的颗粒物中高氯酸盐含量为39.2 ng/m3,PM10的颗粒物质含量为9.89 ng/m3。
某些农业活动也会带来水体的高氯酸盐污染,可能是由于施用某些农业肥料中含有高氯酸盐从而引起污染。如Backus M S等[23]监测的采集自Great Lakes Basin的55个水样中有少量点位检出高氯酸盐(0.20 ~0.33 μg/L,检出限为 0.20 μg/L),其推测原因之一即为农业肥料施用引起。
而对于该次监测,经调查,在河流Ⅲ和Ⅳ的上游支流、干流(断面7#下游)存在多家高氯酸盐生产企业,由于生产工艺的限制,部分企业的废水、甚至周边空气中均存在不同浓度氯酸盐。经监测发现,这些企业外排废水中高氯酸盐含量在几每升毫克、几十每升毫克、甚至几千每升毫克浓度水平,其下游1 km河流中高氯酸盐浓度高达几十至几百每升毫克不等。由于中国目前尚无相关的高氯酸盐环境质量标准和排放标准,这些废水中的高氯酸盐未经处理直接排入河流中,引起高氯酸盐污染。河流Ⅴ则主要是由于位于河流Ⅲ和Ⅳ的下游而含有一定浓度的高氯酸盐。有大量以高氯酸钾为直接产品的化工厂,广泛分布在全国各地。在工业生产中利用高氯酸铵作为氧化添加剂的更是普遍。同时中国还是烟火制造和消费大国,烟火中氧化剂的主要成分就是高氯酸钾。更值得一提的是,2002年国务院下达52号文明令禁止采用氯酸钾配置烟火,不少厂家却用高氯酸钾作为了氯酸钾的替代物,从而大大提高了高氯酸盐的生产和使用量。
该次调查还发现,一般情况下的自然水体中应该不含有高氯酸盐(笔者曾对取自雪水融化汇集而成的小溪流样品进行监测,未检出高氯酸盐),但由于人类活动,如烟花爆竹的燃放、农药化肥的施用等,即使周边无工业污染源的水体中仍然会检出痕量的高氯酸盐。这也是河流Ⅰ和Ⅱ检出痕量高氯酸盐的主要原因。
3 结论
在已优化的条件下,离子色谱-串联质谱技术测定地表水中高氯酸盐含量的方法,样品经抑制电导检测后通过阀切换技术去除水样中大量弱保留离子,进行负离子电喷雾选择离子监测(m/z=99),实现了直接进样分析水样中痕量的高氯酸盐。方法灵明度高、精密度好、准确度高:检出限达0.031 μg/L;低、中、高浓度样品标准偏差为2.26%~4.45%,加标回收率为 93.0% ~98.0%,与《离子色谱检测饮用水中高氯酸盐》(US EPA Method314.0)方法比对吻合。四川省主要河流高氯酸盐含量范围在未检出(检出限:0.031 μg/L)~68.0 μg/L 之间,除 2 条河流浓度(小于1.30 μg/L)较低外,其余3条河流含量较高,高氯酸盐污染不可忽视。经调查,含量较高的河流污染由上游支流、干流高氯酸盐生产企业废水排放引起。
[1]周勇.水环境中的高氯酸盐污染[J].工程与建设,2007,21(3):335-336.
[2]Trumpolt W C,Crain M,Cullison D G,et al.Perchlorate:Sources,uses,and occurrences in the environment[J].Remediation Journal,2005,16(1):65-89.
[3]Kosaka K,Asami M,Kunikane S.Perchlorate:Origin and occurrence in drinking water[C]//Nriagu J O.Encyclopedia of Environmental Health. USA:Elsevier,2011:371-379.
[4]Charnley G.Perchlorate:Overview ofrisksand regulation[J].Food and Chemical Toxicology,2008,46(7):2 307-2 315.
[5]Baier-Anderson C. Risk assessment, remedial decisions and the challenge to protect public health:The perchlorate case study[J].Analytica Chimica Acta,2006,567(1):13-19.
[6]于佳,唐玄乐,刘家仁.高氯酸盐对人体健康影响的研究进展[J].环境与健康杂志,2008,25(7):648-650.
[7]Park J,Rinchard J,Liu F J,et al.The thyroid endocrine disruptor perchlorate affects reproduction,growth,and survival of mosquitofish[J],Ecotoxicology and Environmental Safety,2006,63(3):343-352.
[8]Greer A M,Goodman G,Pleus C R,et al.Health effects assessment for environmental perchlorate contamination:The dose response for inhibition of thyroidal radioiodine uptake in humans[J].Environmental Health Perspectives,2002,110(9):927-937.
[9]Motzer W E.Perchlorate:Problems,detection,and solutions[J].Environmental Forensics,2001(2):301-311.
[10]Quinones O,Oh J E,Vanderford B,et al.Perchlorate assessment of the Nakdong and Yeongsan watersheds,republic of korea[J].Environ.Toxical.Chem.,2007,26(7):1 349-1 354.
[11]Kannan K, PraamsmaL M, OldiF J, etal.Occurrence of perchlorate in drinking water,groundwater,surface water and human saliva from India[J].Chemosphere,2009,76(1):22-26.
[12]Wu Q,Zhang T,Sun H W,et al.Perchlorate in tap water,ground water,surface waters,and bottled water from China and its association with other inorganic anions and with disinfection byproducts[J].Arch Environ Contam Toxico1,2010,58(3):543-550.
[13]刘晓锋,黄克建,李 璐.离子色谱法检测爆炸残留物中的氯酸根和高氯酸根[J].广西科学院学报,2010,26(3):268-269.
[14]US EPA method 314.0,Determination of perchlorate in drinking water using ion chromatography[S].
[15]叶龙,尤宏,姚杰,等.固相萃取-离子色谱法测定地下水中痕量的高氯酸根离子[J].色谱,2012,30(1):76-79.
[16]US EPA method 314.2,Determination of perchlorate in drinking water using two-dimensional ion chromatography with suppressed conductivity detection[S].
[17]US EPA Method 331.0,Determination of perchlorate in drinking water by liquid chromatography electrospray ionization mass spectrometry[S].
[18]US EPA Method 332.0,Determination of perchlorate in drinking water by ion chromatography with suppressed conductivity and electrosprayionization mass spectrometry[S].
[19]HJ 168—2010 环境监测 分析方法标准制修订技术导则[S].
[20]TikkanenW M. Developmentofdrinkingwater regulation for perchlorate in California[J].Analytica Chimica Acta,2006,567(1):20-25.
[21]Kosaka K,Asami,M,Matsuoka Y.Occurrence of perchlorate in drinking water sources of metropolitan area in Japan[J].Water Research,2007,41(15):3 474-3 482.
[22]hi Y l,Zhang N,Gao J M.Effect of fireworks display on perchlorate in air aerosols during the Spring Festival[J].Atmospheric Environment,2011,45(6):1 323-1 327
[23]Backus S M,Klawuun P,Brown S,Determination of perchlorate in selected surface waters in the Great Lakes Basin by HPLC/MS/MS[J].Chemosphere,2005,61(6):834-843.
