粒子插入法计算一氧化碳在乙醇中的溶解度
2014-04-21宣爱国侯俊烨吴元欣闫志国严亮
宣爱国,侯俊烨,吴元欣,闫志国,严亮
1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程省部共建教育部重点实验室(武汉工程大学),湖北 武汉 430074
粒子插入法计算一氧化碳在乙醇中的溶解度
宣爱国1,2,侯俊烨1,2,吴元欣1,2,闫志国1,2,严亮1,2
1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程省部共建教育部重点实验室(武汉工程大学),湖北 武汉 430074
采用等温等压(NpT)系综结合Widom粒子插入法计算了298、323 K下1~6 MPa压力范围内一氧化碳在乙醇中的溶解度,探讨了压力、温度对一氧化碳溶解度的影响.模拟过程中,一氧化碳采用DREIDING力场,乙醇采用OPLS-ua力场,模拟结果与文献实验值吻合良好.结果表明温度恒定时一氧化碳在乙醇中的溶解度随着压力的增加而增大,且溶液中一氧化碳的摩尔分数与压力的关系满足亨利定律;压力相同时,一氧化碳的溶解度在298、323 K下的差别很小,说明在一定范围内,一氧化碳在乙醇中的溶解度受温度影响不大.
粒子插入法;蒙特卡罗模拟;一氧化碳;乙醇;溶解度
0 引言
气液相平衡数据在化工生产与理论研究中有着重要作用,但其实验测定较为困难.近年来,随着计算机技术的高速发展,分子模拟已经成为除实验测定、模型计算外的一种全新而独特的研究手段[1].分子模拟建立在统计热力学与分子力学理论基础之上,在计算热力学数据的过程中只依赖于描述原子间相互作用的分子力学力场,直接从微观状态分布来求解宏观量,在计算相平衡方面具有巨大优势.
计算体系气液相平衡常用且计算效果较好的方法包括Widom粒子插入法[2]与Gibbs系综蒙特卡罗(Gibbs ensemble Monte Carlo,GEMC)[3-4]方法.前者通过计算插入的虚拟粒子与体系粒子之间作用的超额化学势来计算气体的溶解度或亨利系数,而后者则分别模拟气、液两相,通过加入粒子在两相间的交换移动达到气液相平衡.与GEMC方法相比,粒子插入法在实施过程中更为便捷,且在计算亨利系数方面更为直接.在2004年的第二届工业流体模拟竞赛(the Second Industrial Fluid Property Simulation Challenge,2nd IFPSC)[5]中,Widom方法被用于计算N2、O2、CO2和CH4四种气体在乙醇中的亨利系数.此后,随着力场的不断完善,粒子插入法逐渐被用于多种体系[6-9]气液相平衡的计算,且都取得了令人满意的结果.
一氧化碳、乙醇是羰基化法制备碳酸酯[10-11]的重要原料和溶剂.研究碳酸酯相关合成体系的相平衡可为产物分离方法的选择、分离设备平衡级数的计算及尺寸设计等提供依据.本文采用NpT系综结合粒子插入法,通过Monte Carlo算法计算了不同条件下一氧化碳在乙醇中的溶解度及亨利系数,将计算结果与文献值进行比较,发现粒子插入法可以取得较好的结果.
1 模拟方法
1.1 粒子插入法计算原理
一氧化碳在乙醇中的溶解度较低,溶解了一氧化碳的乙醇可视为无限稀释溶液.溶液中的相互作用可直接视为乙醇分子与一氧化碳分子之间的相互作用,而不考虑液相中气体分子之间的相互作用.故可采用Henry定律来描述一氧化碳-乙醇体系的相平衡关系.
根据溶液热力学,亨利系数H2,1可由溶质在无限稀释溶液中的超额化学势μ2ex,∞来进行计算[12]:

式中,kB是Boltzmann因子,ρ1为溶剂密度,T为体系温度.由此可知,计算亨利系数的关键在于准确地计算溶质的超额化学势μ2ex,∞.采用Widom粒子插入法计算化学势的基本步骤为:在模拟进行过程中冻结一模拟构型,在模拟盒中随机插入一个粒子,计算此粒子与体系中其他粒子相互作用的势能之和,然后将插入粒子撤出,恢复原来的模拟构型,继续进行模拟.采用NpT系综模拟气液相平衡时,插入的溶质粒子的超额化学势μ2ex可表示为[13]:
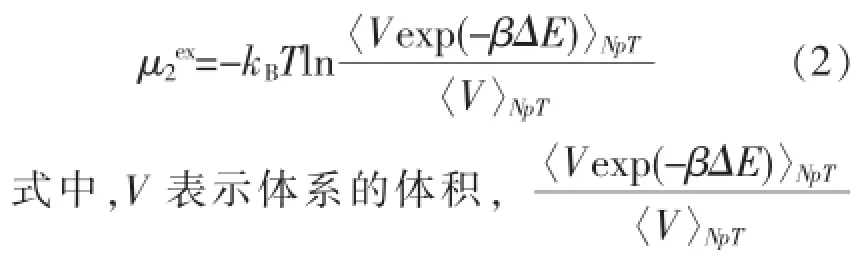
表示对体系能量进行等温等压系综平均.当体系足够大时,即得到溶质在无限稀释溶液下的超额化学势μ2ex,∞.
1.2 分子力场
模拟过程中,溶剂分子乙醇采用OPLS-ua力场,乙醇分子由甲基、亚甲基及羟基氢、氧原子四个作用位点构成,电荷分布在亚甲基及羟基氢、氧原子上,详细力场参数见文献[14].溶质分子一氧化碳采用DREIDING力场,碳、氧原子上分别负载有0.020 3 e的正、负电荷[15],其余力场参数见文献[16].
体系中分子i和j之间的相互作用势能Eij可表示为:

式中,ELJ为Lennard-Jones势能,Echarge为电荷作用部分势能,分别表示为:

式中,na,nb分别为分子a和b的作用位点数;qi,qj分别为作用位点i及j的电荷量;rij为作用位点i与j之间的距离;ε0为真空介电常数.
不同作用位点之间的相互作用采用Geometric混合规则:

式中,εi,σi分别代表作用位点i的LJ势能能量参数与尺寸参数.
2 模拟结果与讨论
采用NpT系综结合Widom粒子插入法计算一氧化碳-乙醇体系的气液相平衡.模拟盒采用周期性边界条件,初始边长设为30 nm,模拟温度分别为298、323 K,压力范围为1~6 MPa.采用球形割去法进行势能截断,截断半径为1 nm,截断距离外的范德华作用通过长程矫正法进行矫正.静电作用通过Ewald加和法进行计算.模拟过程共进行300 000次Monte Carlo(MC)循环,其中,前250 000次为平衡阶段,后50 000次为统计计算阶段.模拟过程的MC移动及其发生的概率分别为:体积波动,1%;偏倚构型增长,32%;质心平移,34%;绕质心的旋转,33%.各移动的接受概率均设为0.5.本文的模拟工作均使用Towhee-7.0.2[17]软件包完成.
2.1 压力对气体溶解度的影响
图1(a)、图1(b)分别表示温度298、323 K时一氧化碳在乙醇中的溶解度的模拟结果与文献结果[18]之间的比较.

图1 一氧化碳在乙醇中的溶解度随压力的变化Fig.1Effect of pressure on solubility of carbon monoxide in ethanol
从图中可知,温度一定时,一氧化碳在乙醇中的溶解度随压力的增大而增加,且溶质在溶液中的摩尔分数与压力呈很好的线性关系,满足亨利定律.在模拟计算采用的压力范围内,溶解度的模拟值与文献值非常接近.在1~4 MPa压力范围内,溶解度模拟值总是略大于文献实验值,但总体均在可接受范围内:298 K下,模拟值与文献值的相对偏差在4.2%~12.9%之间;323 K下,二者的相对偏差在5.5%~18.3%之间.压力大于4 MPa后,由图中拟合模型可知,溶解度的模拟值与模型值较1~4 MPa范围内的值相比更为接近,298 K下模拟值与模型值的相对偏差范围降至2.1%~3.3%;323 K下二者相对偏差范围则降至0.47%~3.9%.
2.2 温度对气体溶解度的影响
图2(a)、图2(b)分别为298~323 K范围内一氧化碳在乙醇中溶解度的文献结果与模拟结果.实验结果表明,压力一定时,一氧化碳在乙醇中的溶解度几乎不随温度的变化而发生改变.压力一定时,模拟得到的溶解度值在298~323 K的温度范围非常相近,最大偏差为0.28%,且两个温度下回归所得的溶解度模型与文献模型有着很相似的趋势,模拟结果证实了实验结果的准确性,同时也证明使用粒子插入法可以预测一定范围内温度对一氧化碳-乙醇体系气液相平衡的影响.
图3为298 K、323 K下一氧化碳-乙醇体系亨利系数的模拟拟合值与文献值之间的对比.

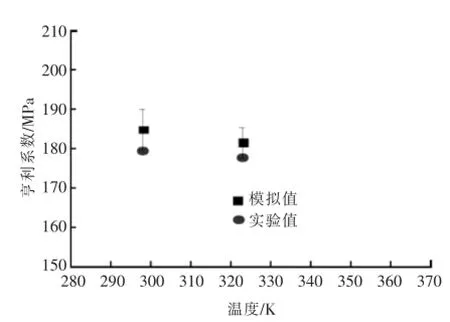
图3 一氧化碳在乙醇中的亨利系数模拟值与文献值之间的对比Fig.3Comparison of Henry’s law constants between simulated and experimental data
模拟与实验结果均表明一氧化碳-乙醇体系的亨利系数在298 K与323 K下几乎可视为相等;且模拟所得的亨利系数与文献结果非常接近,在298、323 K下,二者的相对偏差分别为2.9%、2.1%,说明采用与NpT结合的粒子插入法模拟一氧化碳-乙醇体系亨利系数是可取的,其结果可用于预测本研究范围内其他压力下一氧化碳在乙醇中的溶解度.
3 结语
本研究采用NpT系综结合Widom粒子插入法计算了298、323 K下1~6 MPa范围内一氧化碳在乙醇中的溶解度,计算结果及拟合模型均与文献结果吻合良好,证实了分子模拟可有效地检验与预测实验数据.同时,模拟结果与文献结果均表明:温度恒定时,一氧化碳在乙醇中的溶解度随压力的增加而增大,且二者之间的关系符合亨利定律;一定范围内,温度对一氧化碳溶解度的影响并不明显,一氧化碳在乙醇中的亨利系数几乎不随着温度的变化而改变.
致谢
感谢国家自然科学基金委和乙烯工程下游产品开发及过程强化湖北省协同创新项目组提供的资助.
符号说明
E-势能,J;Hi-溶液中组分i的亨利系数,MPa;kBBoltzmann常数,J·K-1;N-模拟分子总数;n-分子作用位点数;p-压力,MPa;q-作用位点的电荷量,e;R-气体常数,J· mol-1·K-1;rij-作用位点i与j之间的距离,nm;T-绝对温度,K;V-体积,m3;xi-溶液中组分i的摩尔分数.
希腊字母:ε-分子能量参数,J;ε0-真空介电常数,F·m-1;μi-组分i的化学势;ρ-数密度,m-3;σ-分子尺寸参数,nm.
上角标:ex-超额热力学性质;∞-无限稀释值.
[1]朱宇,陆小华,丁皓,等.分子模拟与化学工程[J].化工学报,2004,55(8):1213-1223.
ZHU Yu,LU Xiao-hua,DING Hao,et al.Molecular simulation in chemical engineering[J].CIESC Journal,2004,55(8):1213-1223.(in Chinese)
[2]WIDOM B.Some topics in theory of fluids[J].J.Chem Phys.,1963,39(11):2808-2812.
[3]PANAGIOTOPOULOS A Z.Direct determination of phase coexistence properties of fluids by Monte Carlo simulationinanewensemble[J].Molecular Physics,1987,61(4):813-826.
[4]PANAGIOTOPOULOS A Z,QUIRKE N,STAPLETON M,et al.Phase equilibria by simulation in the Gibbs ensemble alternative derivation,generalization and application to mixture and membrane equilibria[J].Molecular Physics,1988,63(4):527-545.
[5]CASE F,CHAKA A,FRIEND D G,et al.The second industrial fluid properties simulation challenge[J].Fluid Phase Equilibria,2005,236(1):1-14.
[6]吴晓萍,刘志平,汪文川.分子模拟研究气体在室温离子液体中的溶解度[J].物理化学学报,2005,21(10):1138-1142.
WU Xiao-ping,LIU Zhi-ping,WANG Wen-chuan.Molecular dynamics simulation of gas solubility in room temperature ionic liquids[J].Acta Phys.-Chim.Sin.,2005,21(10):1138-1142.(in Chinese)
[7]WU Chuan-jie,LI Xiao-feng,DAI Jian-xing,et al.Prediction of Henry’s law constants of small gas molecules in liquid ethylene oxide and ethanol using forcefieldmethods[J].FluidPhaseEquilibria,2005,236(1):66-77.
[8]HUANG Yow-lin,MIROSHNICHENKO S,HASSE H,et al.Henry’s law constant from molecular simulation:a systematic study of 95 systems[J].International Journal of Thermophysics,2009,30(6):1791-1810.
[9]潘杨.TDI相关合成体系相平衡的测定与分子模拟[D].武汉:武汉工程大学,2013.
PAN Yang.Determination and molecular simulation of TDI related synthesis system[D].Wuhan:Wuhan Institute of Technology,2013.(in Chinese)
[10]田琦峰,张光绪,吴广文,等.碳酸二苯酯的合成研究进展[J].精细石油化工进展,2001,2(9):37-41.
TIAN Qi-feng,ZHANG Guang-xu,WU Guangwen,etal.Researchprogressinsynthesisof diphenyl carbonate[J].Advances in Fine Fetrochemicals,2001,2(9):37-41.(in Chinese)
[11]刘定华,徐明,陈振松,等.铜盐催化乙醇氧化羰基化反应性能的研究[J].高校化学工程学报,2013,27(3):425-430.
LIU Ding-hua,XU Ming,CHEN Zhen-song,et al.Study on reaction performance of copper salt in oxidative carbonylation of ethanol[J].Journal of ChemicalEngineeringofChineseUniversities,2013,27(3):425-430.(in Chinese)
[12]SHING K S,GUBBINS K E,LUCAS K.Henry constants in non-ideal fluid mixtures:Computer simulation and theory[J].Molecular Physics,1988,65(5):1235-1252.
[13]ALLEN M P,TILDESLEY D J.Computer simulationsofliquids[M].Oxford:ClarendonPress,1987:2-240.
[14]JORGENSEN W L.Optimized intermolecular potentialfunctionsforliquidalcohols[J].J.Phys.Chem.,1986,90(7):1276-1284.
[15]SWEATMAN M B,QUIRKE N.Modelling gas adsorption in slit-pores using Monte Carlo simulation[J].Molecular Simulation,2001,27(5-6):295-321.
[16]MAYO S L,OLAFSON B D,GODDARD III W A.DREIDING:A genericforcefieldformolecular simulations[J].Phys Chem.,1990,94(26):8897-8909.
[17]MARTIN M G.MCCCS T owhee:a tool for Monte Carlo molecular simulation[J].Mloecular Simulation,2013,39(14/15):1212-1222.
[18]TONNER S P,WAINWRIGHT M S,TRIMM D L.Solubility of carbon monoxide in alcohols[J].Journal of Chemical and Engineering Data,1983,28(1):59-61.
Solubility simulation of carbon monoxide in ethanol using Widom insertion method
XUAN Ai-guo1,2,HOU Jun-ye1,2,WU Yuan-xin1,2,YAN Zhi-guo1,2,YAN Liang1,2
1.School of Chemical Engineering&Pharmacy,Wuhan Institute of Technology,Wuhan 430074,China;2.Key Laboratory of Green Chemical Process(Wuhan Institute of Technology),Ministry of Education,Wuhan 430074,China
Widom insertion method was used to calculate the solubility of carbon monoxide in ethanol.The simulation was performed under isothermal-isobaric conditions of 298,323 K and 1-6 MPa.Effects of pressure and temperature on solubility of carbon monoxide in ethanol were discussed.During the simulation,the universal force field DREIDING was chosen for solute carbon monoxide,while the unitedatom force field OPLS-ua was chosen for solvent ethanol.The simulated data show good agreement with experimental results.The results indicate that solubility of carbon monoxide in ethanol increases with increasing pressure when the temperature is specified;the relationship between mole fraction of carbon monoxide and pressure can be described by Henry’s Law;however,the solubility of carbon monoxide obtained at 298 K and 323 K shows little difference when the pressure is the same,which means the dissolution of carbon monoxide in ethanol is not sensitive to temperature.
Widom insertion method;Monte Carlo simulation;carbon monoxide;ethanol;solubility
TQ02
A
10.3969/j.issn.1674-2869.2014.012.003
1674-2869(2014)012-0011-05
本文编辑:张瑞
2014-11-08
宣爱国(1957-),女,湖北松滋人,教授,博士,硕士研究生导师.研究方向:化工过程模拟与优化.
