15N示踪法测定海洋反硝化速率中29N2浓度的准确定量
2014-04-18曾健陈敏郑敏芳邱雨生
曾健,陈敏,2*,郑敏芳,邱雨生,2
(1.厦门大学 海洋与地球学院,福建 厦门 361005;2.近海海洋环境科学国家重点实验室,福建 厦门 361005)
1 引言
固氮速率和反硝化速率的相对平衡,决定了海洋无机氮储库的大小,直接影响着海洋生态系统的生物泵效率乃至海洋吸收大气CO2的能力[7]。诸多古海洋沉积记录已证明,在地质历史时期,海洋的固氮作用和反硝化作用相对强弱发生着周期性的变化[8—10],并且与地球气候的波动密切相关[3,11—12]。学术界对现代海洋固氮速率和反硝化速率是否平衡一直存在很大争议[4,13—17],这也使得海洋氮循环成为备受关注的热点之一。目前的研究结果认为,海洋由反硝化作用迁出的结合态氮通量达到400Tg/a左右[15,18—19],而海水的反硝化速率约为100Tg/a[9,18],且主要集中在阿拉伯海和东赤道南、北太平洋等三大氧极小值区[18]。然而,近年来厌氧氨氧化作用及其他新型脱氮过程的发现,极大地改变了有关海洋氮循环的传统认识[20—21],也使得反硝化作用在海洋中的主导地位面临挑战[22—25]。因此,准确评估海洋反硝化作用的强度以及相关的调控因素,有助于深入了解现代海洋的氮循环路径。
基于15N示踪的同位素配对技术(Isotope Pairing Technique,IPT),因其快速、灵敏的特点[26],尤其能够定性和定量区分反硝化和厌氧氨氧化过程而被广泛应用于海洋氮循环研究中[27—30],也是目前最重要的技术手段之一。N2是反硝化作用的最终产物,也是分析测试的对象。由于环境中的空气具有很高的N2含量,因此,避免样品被空气污染是准确定量获得反硝化速率的关键。15N示踪测定反硝化速率的实验过程中,涉及到赶气、水样转移、示踪剂添加、样品存放等一系列流程,这些都很难避免不会受到空气背景的污染[31]。寻找一种能够有效校正空气背景影响的方法,对于准确定量反硝化速率尤为重要。本研究通过对南海海水反硝化速率测定过程中所获得的N2及其同位素组成的数据进行分析,探索测定过程中空气的N2背景是否对实测结果产生了影响,进而提出了校正空气29N2背景影响的方法,通过对实际海水样品的分析,验证所提出校正方法的可行性。
2 材料与方法
2.1 样品采集
2012年8—9月期间,搭载“实验3号”科学考察船,采集南海中心海盆(10°~18°N,110°~118°E)300~1 500m深度区间的海水,运用15N标记示踪的同位素配对技术开展反硝化速率的测定。为避免采样过程中的空气污染,本实验在郑敏芳[32]研究的基础上加以改进,即由CTD-Rossett采水器采集的海水先用乳胶管引出并注入到250cm3玻璃瓶内[33],乳胶管的出水口始终保持在液面下方1cm左右处,待水样溢流3倍采样瓶体积后,旋紧瓶盖,在室温下避光存放。每个层位采集了双份平行样。
2.2 水样的赶气和转移
对每个层位所采集的两份水样分别进行预先赶气和不赶气处理[32]。取其中一份水样,将一支通有高纯He气(纯度99.999%)的15cm长不锈钢针穿过带硅胶垫的瓶盖插至瓶底,同时在硅胶垫上扎一个小针头以排出空气,吹扫30min。将吹扫、赶气后的水样通过高压He气转移至10支预先用高纯He气吹扫过的12.5cm3带橡胶垫密封培养瓶内(Labco Exetainer,Wycombe,UK),顶空部余留4cm3体积。另一份水样不做高纯He预先的赶气处理,以相同的方法转移至10支密封的培养瓶内。所采用的这种培养瓶也是目前国内外研究中最为广泛采用的培养瓶[23—30,32—33]。
2.3 水样的培养实验
用微量进样针将0.06μmol K15NO3(15N 原子丰度98.50%,上海化工研究院)溶液加入各培养瓶内,使得培养体系中15N的丰度约为20%,此过程培养瓶始终保持密封。每个层位的水样均设置5个培养时间段,即0h、48h、72h、96h和120h,每个时间段的样品设置了双份平行样。在培养的终点,用10μm3饱和HgCl2溶液抑制生物的活性。所有样品均在采样后3h之内完成培养前的处理,且都在室温下的无光的水槽中进行。培养结束后,样品倒置在室温水槽内密封存放,带回陆地实验室进行N2浓度及其同位素组成的分析。
2.4 N2及其同位素组成的质谱分析
采用Gas BenchⅡ自动进样器与DELTAPlusXP稳定同位素比值质谱仪(Thermo Finnigan,USA)联机方式对样品 的28N2、29N2和30N2同 时 进行测定。分析测试前,先对样品进行加热(40℃)超声处理40min[32],将溶解在海水中的N2完全驱赶至培养瓶的顶空部,之后进样进行测定。样品从采集到分析间隔1个月左右的时间。分析时采用空气为标准进行测量,本研究中N2浓度测定的相对标准偏差小于8%[32]。
2.5 反硝化速率的计算
在一个仅存在反硝化作用的培养体系中,加入15N示 踪 物 后,会 产 生28N2、29N2和30N23种质量数的N2。假设反硝化细菌随机利用体系中的14N和15N,那么由生物产生的这3种分子之间的比例关系与溶液中15N的丰度(即15N浓度占体系N总浓度的份额,用FN表示)有关[32,34],可表示为:

因此,通过测定29N2、30N2分子的产生速率,即可计算反硝化速率[33,35]:
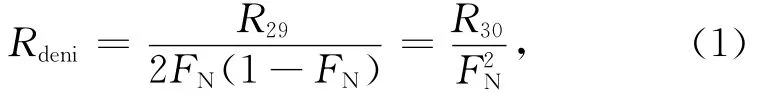
其中,Rdeni、R29和R30分别代表反硝化速率、29N2产生速率以及30N2产生速率。后两者可通过对不同培养时间 段 产 生 的29N2和30N2浓 度 做 线 性 拟 合 得到[23—24]。
3 结果与讨论
3.1 29N2、30N2与28N2峰面积的比较
将所有实验数据的29N2峰面积(Area 29)和30N2峰面积(Area 30)分别对28N2峰面积(Area 28)作图,发现这些数据点均落在一条直线的上方。若进一步将这些数据分为3组:(1)未经赶气处理且不存在反硝化作用的样品;(2)经过赶气处理但不存在反硝化作用的样品;(3)存在反硝化作用的样品,则可明显看出,前两组样品的数据都落在一条直线上,而存在反硝化作用样品的数据则离散地分布在直线上方(图1)。对于不存在反硝化作用的样品,将未经赶气和经赶气处理的Area 29和Area 28分别进行拟合,可得其关系分别为y=0.007 29x+0.003(r2=0.998 9,n=34)和y=0.007 35x+0.000 04(r2=0.999 9,n=152);同样地,对这两组样品 Area 30和 Area 28进行线性拟合得到的结果分别为y=0.003 44x-0.021(r2=0.920 0,n=34)和y=0.002 81 x-0.004(r2=0.971 1,n=152)。上述拟合结果说明,在没有反硝化作用发生的情况下,水样中所溶存氮气的29N2/28N2组成相当恒定,且赶气和未经赶气处理的样品之间29N2/28N2组成几乎没有差异(偏差小于1%),而未经赶气处理的样品30N2/28N2比值比赶气处理的30N2/28N2比值来得高,相对偏差大于20%。此前不同学者对采自全球不同区域的大气样品进行过分析,结果表明,空气中 N2的15N/14N 比 值 几 乎 恒定。Junk和Svec对5份来自全球不同海拔地区的空气样品进行了 分 析,其29N2/28N2比 值 平 均 为 0.007 351±0.000 001[36];Mariotti于不同季节对环法国17个地区的空气样品进行分析后指出,δ15N标准偏差小于0.03×10-3,即15N相对丰度的偏差小于1×10-5[37],说明大气的15N/14N比值随时间和空间的变化很小。Berglund和Wieser为国际纯粹与应用化学联合会(IUPAC)撰写的“元素同位素组成2009”报告中进一步确定了大气中15N相对丰度最理想的测试结果为0.366 3%,换 算 成29N2/28N2的 理 论 比 值 即 为0.007 35[38]。结合本研究结果,可以确定空气中29N2/28N2的比值恒定为0.007 35。
3.2 水样中N2和30N2峰面积过剩的来源分析

图1 29 N2、30 N2 和28 N2 峰面积的关系
空 气 中15N 的 丰 度 为 0.366%[39—41]。假 设 N2由15N和14N原子随机组合形成,根据理论计算,空气中29N2/28N2的比值应为0.007 35,而30N2/28N2的比值应为0.000 013 5。比较发现,不存在反硝化作用的样品中溶解气体的29N2/28N2比值与空气的29N2/28N2理论比值几乎完全一致,但不存在反硝化作用实测样品的30N2/28N2比值却较空气的理论值高出2个数量级。在一个15N丰度为20%的培养体系中,若存在反硝化作用,假设细菌随机利用14N和15N,且不考虑同位素分馏效应,那么实际产生的29N2/28N2和30N2/28N2比值应分别为0.5和0.062 5,均远大于上述实测值。若培养体系中存在厌氧氨氧化作用,由于该过程不产生30N2,故会导致30N2/28N2比值的降低,而不是升高[27]。基于上述考虑,可以排除生物过程产生过剩30N2的可能性。
为进一步证实这些水样溶解的N2来自于大气,取以往不同时间利用实验周围空气所做的标准工作曲线进行对比,可发现实测得的29N2/28N2比值平均为(0.007 33±0.000 03),与理论值几乎吻合;而实测得的30N2/28N2比值平均为(0.001 50±0.000 51),同样较理论值高出2个数量级,但略低于上述样品的测定值(图2)。由此可以说明,研究样品中确有空气残留的N2存在,而且这部分来自于空气的N2具有30N2显著“过剩”的特征。水样中残留的N2可能由于赶气不充分或者是样品存放过程中外界空气的扩散进入。郑敏芳[32]曾仔细探索过赶气过程去除培养瓶N2的效率,结果表明当赶气时间超过10min后,培养瓶内N2浓度低于检测限,因此水样中残留的N2更可能是在样品长时间存放期间外界空气的扩散进入。由于实测空气的29N2/28N2比值与样品测得的29N2/28N2比值十分接近,说明在外界空气扩散进入培养瓶时,并未导致28N2和29N2的明显分馏。
如此,样品和空气中“过剩”30N2的存在是否是其它组分干扰的结果?30N2被质谱的离子源轰击后产生30N离子,核质比m/e为30。近些年的研究表明,N2O分子被质谱离子源的电子轰击后会产生NO+碎片,且与N2O+离子个数有恒定的比例关系[42]。氮、氧 元 素 分 别 以14N(99.637%)和16O(99.757%)丰度最高,故 m/e为30的14N16O+是主要碎片。空气中 N2O的含量约为300×10-9[43],与 N2含量的比值为4×10-7;海水中溶解N2O平均浓度小于50nmol/kg[43],仅相当于溶解 N2浓度(500μmol/kg)的万分之一,显然,即使样品或空气中存在N2O,也无法支持所观察到的30N2过剩现象。除N2O以外,天然海水或空气中存在的分子量为30的气体还包括14N16O、13C17O和12C18O,但它们的含量均远小于N2含量的百万分之一[40],也不可能是产生30N2过剩的主要原因。就目前认识,尚无法清晰界定所观察到的30N2过剩来源,有待后续更深入的研究。鉴于30N2存在未知来源的额外影响,本研究在反硝化速率的计算中仅采用29N2测值进行计算,同时重点考虑培养体系中空气29N2背景的校正方法。

图2 不同时间空气 N2 中29 N2、30 N2 和28 N2 的关系
3.3 29N2峰面积测定结果的校正
根据上述讨论可知,经过培养的水样或者由于赶气不够充分,或是因为存放期间外界空气的扩散进入,导致样品中产生了N2背景,这势必会对反硝化速率的计算结果产生影响。幸运的是,这部分N2具有恒定不变的29N2/28N2比值,故可利用这一特征信号,对实测的29N2峰面积进行校正,从而准确获得经反硝化作用产生的29N2峰面积。
根据空气N2和反硝化作用所产生N2的29N2/28N2比值特征,可建立两种方法来区分反硝化作用与空气对N2的贡献:
方法一:培养后水样中的N2包含生物反硝化作用产生和空气N2背景两部分。设由生物反硝化作用产生的28N2、29N2峰面积分别为a1、b1,空气 N2背景的28N2、29N2峰面积分别为a2、b2;实测的28N2和29N2峰面积分别为A和B。由于空气N2背景中28N2占绝对主导地位,故可假设生物反硝化作用产生的28N2相比于背景可忽略(即a1=0)。另外,由上述讨论可知,b2/a2为一恒定比值,用β表示(β=0.007 35)。由此,可获得如下质量平衡关系:
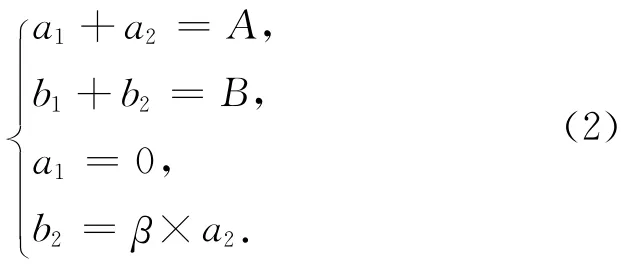
求解b1得:

方法二:若考虑生物作用贡献的28N2,在不存在厌氧氨氧化作用的条件下,假设反硝化细菌在培养体系中随机利用14N和15N,且忽略同位素分馏效应,那么由反硝化作用产生的29N2/28N2峰面积比值等于2FN/(1-FN)[34],用α表示,FN为体系中15N/14N丰度比。同样,可建立如下质量平衡方程:
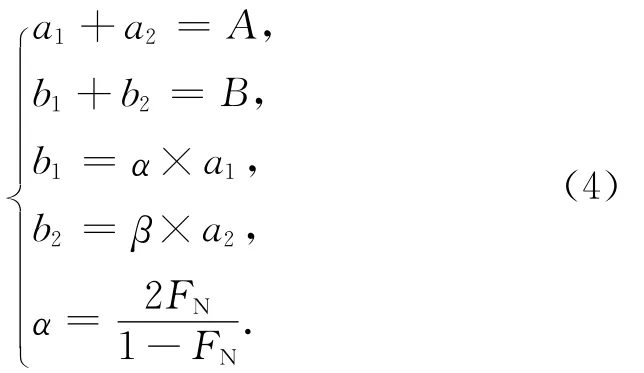
求解b1得:

将两种校正方法得到的b1值进行对比,发现二者所得数据十分吻合(图3)。这个结果也恰好说明上述两种方法所对应的假设前提在实际培养体系中可以成立,即:(1)由生物反硝化作用产生的28N2与空气的28N2背景相比可以忽略;(2)研究样品不存在厌氧氨氧化作用;(3)反硝化作用的同位素分馏效应在计算过程中可以忽略。因此,对于只存在反硝化作用的天然水体,利用15N标记示踪法测定反硝化速率时,可借助式(3)校正空气N2背景的影响。
在此前的研究中,一些学者有考虑到环境本底对29N2浓度测定结果的影响。Dalsgaard等和Nielsen都在各自的研究中提到关于“过剩29N2”的概念,即生物过程产生的29N2[30,34],但对计算的方法并未给出具体说明。Holtappels等提出了修正29N2背景干扰的具体方法[33],但并未给出背景29N2/28N2的特征比值,也没有考虑厌氧氨氧化作用的存在是否会对校正结果产生影响。样品受空气污染程度与样品保存的方式和时间长短有关。多数国外研究并未涉及29N2背景校正这一方面,可能与样品的存放方式或存放时间有关。即便如此,一个合适的校正方法对获得准确的反硝化速率仍就十分重要[33],这也是本研究提出可以准确校正15N示踪所得反硝化速率的初衷所在。
3.4 29N2校正前后反硝化速率的对比
选取本航次分别位于南海海盆北部和南部的两个站位 KJ12(18°15.12′N,114°0.12′E)和 KJ63(9°59.94′N,111°0.12′E),以氧极小值层位(分别为1 500 m和800m)经预先赶气后水体的培养结果进行对比。当测定的数据未经校正处理时,可以明显地看出平行样的结果之间存在较大的偏差,且29N2浓度与培养时间之间不存在显著的线性相关性(p>0.1)(见图4)。这可能是由于不同培养瓶在放置过程中受外界空气扩散影响的程度不同所致,导致29N2浓度随培养时间增加的信号被背景干扰所掩盖,由此计算得到的反硝化速率将存在很大的不确定度(RSD>50%)。相比之下,对空气29N2背景进行校正后,平行样之间的偏差明显减小,且29N2浓度与培养时间的线性相关性水平显著增加(r2>0.7,p<0.001)(图4),与文献报道的反硝化作用存在证据相符[23-24,33]。经空气29N2背景校后得到的反硝化速率不确定度也显著降低(RSD约为20%)。
就所研究的KJ12和KJ63站氧极小值层位样品,由式(3)进行空气29N2背景校正,并由式(1)计算出的反硝化作用潜在速率平均为(1.8±0.2)μmol/(dm3·d)。该数值落在文献报道的开阔大洋水体反硝化速率范围[0.000 24~2.7μmol/(dm3·d)]之内[24,28]。

图4 KJ12站(a)和KJ63站(b)氧极小值层水样29 N2浓度随培养时间的变化
4 结论
(1)利用15N示踪法测量海水反硝化速率的过程中,样品长时间的存放有可能会受到空气扩散进入的影响,由此导致29N2浓度的分析结果异常,进而影响反硝化速率的准确定量。
(2)由于空气中29N2/28N2比值恒定为0.007 35,并且在空气扩散进入培养体系的过程中该比值几乎不会发生变化,故提出利用该恒定的比值,通过质量平衡方程校正空气29N2背景的影响,在培养体系只存在反硝化作用的情况下,将样品实测的29N2浓度扣除由外界空气贡献的29N2浓度,就可得到由生物反硝化作用产生的29N2准确值,进而可获得准确的反硝化速率。
(3)对南海实际海水样品的研究表明,经空气29N2背景校正后,培养体系29N2浓度与培养时间之间的线性关系显著加强,由此计算出的反硝化速率更为可靠。
[1] Codispoti L A.Phosphorus vs.nitrogen limitation of new and export production[C]//Berger W H.Productivity of the Ocean:Present and Past.New York:Wiley,1989:377-394.
[2] Tyrrell T.The relative influences of nitrogen and phosphorus on oceanic primary production[J].Nature,1999,400(6744):525-531.
[3] Gruber N.The marine nitrogen cycle:overview and challenges[M]//Capone D G,Bronk D A,Mulholland M R,et al.Nitrogen in the Marine Environment.2nd ed.Burlington:Academic Press,2008:1-50.
[4] Gruber N,Sarmiento J L.Global patterns of marine nitrogen fixation and denitrification[J].Global Biogeochemical Cycles,1997,11(2):235-266.
[5] Zehr J P,Kudela R M.Nitrogen cycle of the open ocean:from genes to ecosystems[J].Annual Review of Marine Science,2011,3:197-225.
[6] Knowles R.Denitrification[J].Microbiological Reviews,1982,46(1):43-70.
[7] Falkowski P G.Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2in the ocean[J].Nature,1997,387(6630):272-275.
[8] Ganeshram R S,Pedersen T F,Calvert S E,et al.Large changes in oceanic nutrient inventories from glacial to interglacial periods[J].Nature,1995,376(6543):755-758.
[9] Altabet M A.Isotopic tracers of the marine nitrogen cycle:present and past[M]//Volkman J K.Marine Organic Matter:Biomarkers,Isotopes and DNA.Berlin Heidelberg Springer-Verlag,2006:251-293.
[10] Altabet M A.Constraints on oceanic N balance/imbalance from sedimentary15N records[J].Biogeosciences,2007,4(1):75-86.
[11] Mcelroy M B.Marine biological controls on atmospheric CO2and climate[J].Nature,1983,302(5906):328-329.
[12] Altabet M A,Francois R,Murray D W,et al.Climate-related variations in denitrification in the Arabian Sea from sediment15N/14N ratios[J].Nature,1995,373(6514):506-509.
[13] Codispoti L A,Brandes J A,Christensen J P,et al.The oceanic fixed nitrogen and nitrous oxide budgets:moving targets as we enter the anthropocene?[J].Scientia Marina,2001,65(S2):85-105.
[14] Capone D G,Knapp A N.Oceanography:a marine nitrogen cycle fix?[J].Nature,2007,445(7124):159-160.
[15] Codispoti L A.An oceanic fixed nitrogen sink exceeding 400Tg N a-1vs the concept of homeostasis in the fixed-nitrogen inventory[J].Biogeosciences,2007,4(2):233-253.
[16] Devries T,Deutsch C,Primeau F,et al.Global rates of water-column denitrification derived from nitrogen gas measurements[J].Nature Geoscience,2012,5(8):547-550.
[17] Eugster O,Gruber N.A probabilistic estimate of global marine N-fixation and denitrification[J].Global Biogeochemical Cycles,2012,26(4),doi:10.1029/2012GB004300.
[18] Devol A H.Denitrification including anammox[M]//Capone D G,Bronk D A,Mulholland M R,et al.Nitrogen in the Marine Environment.2nd ed.Burlington:Academic Press,2008:263-301.
[19] Naqvi S W A,Voss M,Montoya J P.Recent advances in the biogeochemistry of nitrogen in the ocean[J].Biogeosciences,2008,5(4):1033-1041.
[20] Zehr J P,Ward B B.Nitrogen cycling in the ocean:new perspectives on processes and paradigms[J].Applied and Environmental Microbiology,2002,68(3):1015-1024.
[21] Brandes J A,Devol A H,Deutsch C.New developments in the marine nitrogen cycle[J].Chemical Reviews,2007,107(2):577-589.
[22] Ward B B.Significance of anaerobic ammonium oxidation in the ocean[J].Trends in Microbiology,2003,11(9):408-410.
[23] Hamersley M R,Lavik G,Woebken D,et al.Anaerobic ammonium oxidation in the Peruvian oxygen minimum zone[J].Limnology and Oceanography,2007,52(3):923-933.
[24] Hannig M,Lavik G,Kuypers M M M,et al.Shift from denitrification to anammox after inflow events in the central Baltic Sea[J].Limnology and Oceanography,2007,52(4):1336-1345.
[25] Dalsgaard T,Thamdrup B,Farías L,et al.Anammox and denitrification in the oxygen minimum zone of the eastern South Pacific[J].Limnology and Oceanography,2012,57(5):1331-1346.
[26] Groffman P M,Altabet M A,Böhlke J K,et al.Methods for measuring denitrification:diverse approaches to a difficult problem[J].Ecological Applications,2006,16(6):2091-2122.
[27] Dalsgaard T,Canfield D E,Petersen J,et al.N2production by the anammox reaction in the anoxic water column of Golfo Dulce,Costa Rica[J].Nature,2003,422(6932):606-608.
[28] Devol A H,Uhlenhopp A G,Naqvi S W A,et al.Denitrification rates and excess nitrogen gas concentrations in the Arabian Sea oxygen deficient zone[J].Deep-Sea ResearchⅠ,2006,53(9):1533-1547.
[29] Bulow S E,Rich J J,Naik H S,et al.Denitrification exceeds anammox as a nitrogen loss pathway in the Arabian Sea oxygen minimum zone[J].Deep-Sea ResearchⅠ,2010,57(3):384-393.
[30] Dalsgaard T,De Brabandere L,Hall P O J.Denitrification in the water column of the central Baltic Sea[J].Geochimica et Cosmochimica Acta,2013,106:247-260.
[31] De Brabandere L,Thamdrup B,Revsbech N P,et al.A critical assessment of the occurrence and extend of oxygen contamination during anaerobic incubations utilizing commercially available vials[J].Journal of Microbiological Methods,2012,88(1):147-154.
[32] 郑敏芳.海洋水体反硝化作用和厌氧氨氧化作用的15N示踪研究[D].厦门:厦门大学,2010.
[33] Holtappels M,Lavik G,Jensen M M,et al.15N-labeling experiments to dissect the contributions of heterotrophic denitrification and anammox to nitrogen removal in the OMZ waters of the ocean[M]//Klotz M G.Research on Nitrification and Related Processes,Part A.Burlington:Academic Press,2011:223-251.
[34] Nielsen L P.Denitrification in sediment determined from nitrogen isotope pairing[J].FEMS Microbiology Letters,1992,86(4):357-362.
[35] Seitzinger S P,Nielsen L P,Caffrey J,et al.Denitrification measurements in aquatic sediments:a comparison of three methods[J].Biogeochemistry,1993,23(3):147-167.
[36] Junk G,Svec H J.The absolute abundance of the nitrogen isotopes in the atmosphere and compressed gas from various sources[J].Geochimica et Cosmochimica Acta,1958,14(3):234-243.
[37] Mariotti A.Atmospheric nitrogen is a reliable standard for natural15N abundance measurements[J].Nature,1983,303:685-687.
[38] Berglund M,Wieser M E.Isotopic compositions of the elements 2009(IUPAC Technical Report)[J].Pure and Applied Chemistry,2011,83(2):397-410.
[39] Montoya J P.Nitrogen stable isotopes in marine environments[M]//Capone D G,Bronk D A,Mulholland M R,et al.Nitrogen in the Marine Environment.2nd ed.Burlington:Academic Press,2008:1277-1302.
[40] Hoefs J.Stable isotope geochemistry[M].6th ed.Berlin Heidelberg:Springer-Verlag,2009:48-58.
[41] 黄达峰,罗修泉,李喜斌,等.同位素质谱技术与应用[M].北京:化学工业出版社,2006:21-35.
[42] Toyoda S,Yoshida N.Determination of nitrogen isotopomers of nitrous oxide on a modified isotope ratio mass spectrometer[J].Analytical Chemistry,1999,71(20):4711-4718.
2
2
3
