PS/PETG 共混物的流变性能和发泡行为研究
2014-04-13刘本刚王向东汪文昭杜中杰
刘本刚,董 帅,王向东,刘 燕,汪文昭,杜中杰,张 晨*
(1.北京化工大学碳纤维及功能高分子教育部重点实验室,北京100029;2.北京工商大学材料与机械工程学院,轻工业塑料加工应用研究所,北京100048;3.中国石油天然气管道局第五工程公司,河北 任丘,062550)
0 前言
一些PS共混物由于能改善PS的流变性能和发泡行为,越来越受到人们的关注。Wang等[1]制备了PS/液晶聚合物(LCP)共混物,表明LCP 的加入能够起到异相成核作用。Zhai等[2]制备了聚丙烯(PP)/PS的共混物,结果表明,随着相容剂份数的增加,PP 与PS之间的相容性变好,两相界面处由于成核能垒低,而更易于发生泡孔成核。随着相容性变好,分散相界面变多,成核密度增加。Huang等[3]制备了不同比例的PP/PS共混物,研究了PP/PS共混物的相态结构及其发泡行为的影响。结果表明,CO2在PP 的溶解度较PS的溶解度高,引入PP 能够提高整个共混物的对CO2的溶解度。
PETG 是典型的极性聚合物,笔者前期的研究表明,PETG 含量和共混物加工工艺对相态结构影响明显[4],本文研 究 了 不 同 含 量 的PETG 对PS/PETG 共混物的流变性能;并对PS/PETG 共混物的发泡行为进行了研究,对泡孔形态和泡孔密度等指标进行了分析,并分析了影响泡孔形态的影响因素。
1 实验部分
1.1 主要原料
PS,158K,扬子石化-巴斯夫有限责任公司;
PETG,SK2008,韩国SK 集团;
CO2,纯度99.5%,北京天罡气体公司。
1.2 主要设备及仪器
双螺杆挤出机,CTE35 ,科倍隆科亚机械有限公司;
高速混合机,GH-10,北京塑胶机械厂;
密炼机,LH-60,上海科创公司;
压片机,LP-S-50,瑞士Labtech公司;
恒温干燥箱,DHG-9425,上海一恒科技有限公司;
旋转流变仪,MARS 2000,美国TA 公司;
扫描电子显微镜(SEM),TESCAN YEGA 11,捷克TESCAN 公司;
真密度计/开闭孔率测定仪,ULTRAPYC 1200e,美国Quantachrome公司;
高压发泡釜,结构如图1所示,自制。
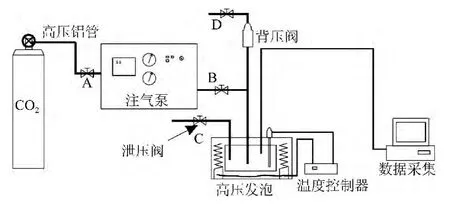
图1 发泡釜的结构Fig.1 Structure of foaming kettle
1.3 样品制备
使用恒温烘箱,将PS、PETG 在60 ℃下烘干2h;按照表1的配方,使用高速混合机将PS和PETG 高速混合5min,然后采用双螺杆挤出机挤出造粒,挤出机1至6区的温度设定为:175、180、185、190、195、200 ℃,机头温度为180 ℃,螺杆转速为100r/min,产品经在线切粒得到PS/PETG 共混物;
采用压片机将PS和PS/PETG 共混物压成2mm后的薄片,用于测定流变性能和进行釜压发泡;
将样品放入高压发泡釜中,使用CO2冲洗高压发泡釜3 min,注入CO2,压力为20 MPa,170 ℃恒温2h,降温至100 ℃,恒温1h,然后快速将釜内压力降至大气压,获得发泡样品。
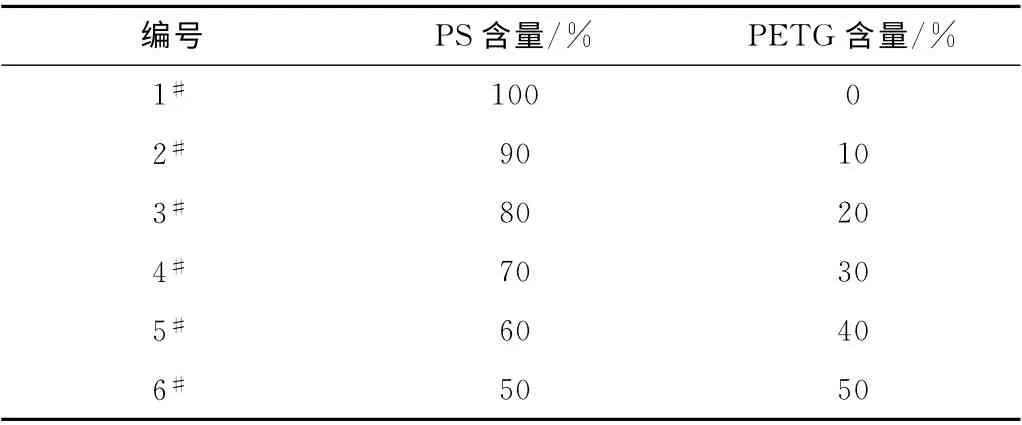
表1 实验配方Tab.1 Formula
1.4 性能测试与结构表征
采用旋转流变仪对PS、PETG 和PS/PETG 共混物的剪切流变性能进行研究,取直径为25mm,厚度为2mm 的试验样品至于平行板间,测试温度为190℃,剪切频率范围为0.1~100s-1;
采用真密度计/开闭孔率测定仪对PS/PETG 共混物发泡样品的密度进行测试,每组样品测试3次,取平均值;
将发泡样品在液氮中冷冻、脆断,表面进行喷金,采用SEM 观察共混物相态结构,加速电压为10kV,在不同放大倍率下观察泡孔形态,泡孔密度计算通过计算机软件Image Tool分析SEM 照片,通过式(1)[5]计算泡沫的泡孔密度,根据式(2),式(1)可简化为式(3)。

式中 N0——泡孔密度,个/cm3
n——统计面积中的泡孔数量,个
A——电镜照片中所选择的统计面积,cm2
Vf——发泡材料中气泡所占比例
ρf——发泡材料的密度,g/cm3
ρ——未发泡材料的密度,g/cm3
2 结果与讨论
2.1 PS/PETG 共混物的流变性能
动态剪切流变仪测量得到的剪切储能模量(G′)能够反映共混物的熔体弹性,是衡量共混物可发性的重要指标之一。G′越高,共混物的熔体弹性也就越好,共混物熔体所具有的熔体强度也就越高,可发性越好。由动态剪切流变测量的损耗因子(tanδ)是损耗角δ 的正切值,是熔体在交变应力的作用下,应变和所受到的应力相位差,tanδ 值越小,相位差越小,共混物的弹性响应越快,黏性耗散越不明显,可发性越高。
从图2中可以看出,在低频区PS/PETG 共混物的G′略高于纯PS 的G′。并且在相同剪切频率下,随着PETG 含量的增加,其相态结构越来越大[4],PS/PETG共混物的G′也在小幅增加。由此可见加入PETG 后,能够提高PS的熔体弹性,从而提高熔体强度,改善PS的可发性。这主要是由于分散相颗粒在受到剪切的情况下,发生的形变松弛过程造成的,Souza等[6]研究了PP/PE-HD 共混物的流变性能,发现PP/PE-HD 共混物的G′要高于纯的PP 和PE-HD,Yee等[7-8]研究了PMMA/PS共混物的流变性能,也发现了同样的现象。
从图3中可以看出,随着剪切频率的增加,纯PS、PETG 和PS/PETG 共混物的tanδ不断下降。在低频区内当剪切频率一定时,PS/PETG 共混物的tanδ 值略高于纯PS,说明其弹性响应较快,黏性耗散现象比较弱,可发性提高。而加入PETG 后,在低频区内的PS/PETG 共混物的tanδ值出现下降,并且随着PETG含量的增加而下降,这与G′的变化原因是一致的。

图2 PS/PETG 共混物的G′Fig.2 Storage modulus of PS/PETG blends

图3 PS/PETG 共混物的tanδFig.3 tanδof PS/PETG blends

图4 PS和不同PETG 含量PS/PETG 共混物泡沫的SEM 照片Fig.4 SEM for foams of PS and PS/PETG blends with different PETG content
2.2 PS/PETG 共混物的发泡行为
采用Image Pro软件对PS 和PS/PETG 泡沫 的SEM 照片进行测量,并使用Origin软件对测量的尺寸进行统计,得到泡孔尺寸的分布图如图5 所示。从图4、图5和图6可以看出,将PETG 引入到PS中,可以明显降低泡孔的尺寸,纯PS泡沫的平均泡孔直径为22.68μm,且泡孔直径分布较宽,泡孔直径的范围从10μm 到35μm 都 有,其 泡 孔 密 度 仅 为3.45×108个/cm3,而加入10%PETG 后,其平均泡孔直径降至7.58μm,泡孔密度提高到4.47×109个/cm3,泡孔直径分布明显变窄,所有泡孔的直径都在5~15μm 的范围内,且其中77%的泡孔直径都在5~10μm 的范围内。继续增加PETG 的用量,泡孔尺寸开始增大,泡孔分布开始变宽。泡孔平均直径增大,而在PETG 含量为50%时,共混物的泡沫则呈现出各相单独发泡的现象,但其平均泡孔尺寸仍然低于纯PS泡沫的平均泡孔尺寸。
出现上述现象主要是由于纯PS发泡过程中,体系是明显的均相成核,均相成核发生时,足够数量的溶解气体分子簇形成一个临界的气泡半径以跨越阻力区,体系的热力学不稳定性是均相成核的驱动力,如图7所示。根据Colton[9-11]的经典成核理论,均相成核速率的表达式如式(4)~(6)所示。

图5 PS和不同PETG 含量PS/PETG 共混物泡沫尺寸分布图Fig.5 Cell size distribution for foams of PS and PS/PETG blends with different PETG content

图6 PS和不同PETG 含量PS/PETG 共混物泡沫的泡沫参数Fig.6 Parameter of PS and PS/PETG blends foam with different PETG content

图7 异相成核与均相成核活化能的对比Fig.7 Activation energycomparison between the heterogenous and homogenous bubble nucleation

式中 f0——气体分子进入临界气泡核的速率因子
C0——气体分子的浓度
ΔP——过饱和压力
Psat——气体在熔体中的饱和压力
Ps——成核发生时的压力,通常Ps为大气压
k——玻尔兹曼常数
T——绝对温度
σ——聚合物―气泡的界面张力
如果在聚合物基体中存在细小的粒子,则可以帮助气泡的形成,成核发生在固体和熔体的界面,界面可以作为成核的催化剂,降低了需要达到一个稳定气泡核的活化能,这种成核方式,称之为异相成核。异相成核是添加成核剂的聚合物发泡体系中最常见的成核方式,从图7可以看出,异相成核可以有效降低用于形成临界气泡核的自由能。异相成核中气泡的生成效率取决于成核剂的种类、形状,固体―气体和固体―熔体的界面张力等。Uhlmann 等[12]给出了异相成核的动力学和数学分析,引入了一个异相成核因子校正均相成核的活化能,如式(7)和(8)所示。
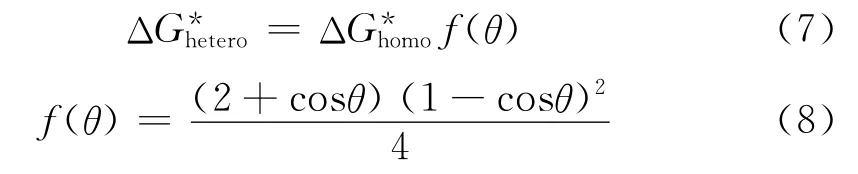
式中 θ——润湿角
f(θ)——异相成核因子
根据Colton等[9-11]的研究,式(6)中的润湿角θ一般为20°,因此在相同的压力降情况下,异相成核所需的活化能较低,成核点能够很容易地产生比均相成核多几个数量级的气泡。
对比图4(a)和图4(b)可以看出,加入PETG 后,PETG 作为分散相起到了明显的异相成核作用,随着PETG 含量的增加,PS/PETG 共混物泡沫的平均泡孔尺寸增大,泡孔密度降低,这主要是由于随着PETG 含量的增加,分散相的尺寸增大,分散相颗粒密度降低,导致异相成核点减少造成的,笔者前期对共混物的研究也证明了这一点[4],当PS/PETG=50/50时,两相形成双相连续的相态,由于在各相中没有另外一种组分作为异相成核点,这是在两相内部都是均相成核,从而造成泡孔平均尺寸的增高和泡孔密度的降低,但在两者的界面上仍存在异相成核的现象,从图4(f)的SEM图上可以明显看出这一现象,所以PS/PETG=50/50时,其平均泡孔尺寸仍低于纯PS,而泡孔密度高于纯PS。
从图4可以看出,加入PETG 后泡沫密度先下降后升高,在PETG 含量为10 %时密度最低,达到0.31g/cm3,而泡孔密度则达到了,4.47×109个/cm3,而纯PS 的密度虽然与加入10 % 时接近,为0.32g/cm3,但是泡孔密度却只有3.45/cm3,说明在使用聚合物共混物发泡时既能获得微孔泡沫塑料,又不会造成泡沫密度的显著上升。而随着PETG 含量的增加,虽然其储能模量会增加,但是由于发泡温度较低,过高的弹性响应会限制泡孔的增长,而在成核密度降低的情况,泡孔尺寸不能够显著增加时,直接结果就是泡沫密度的提高。
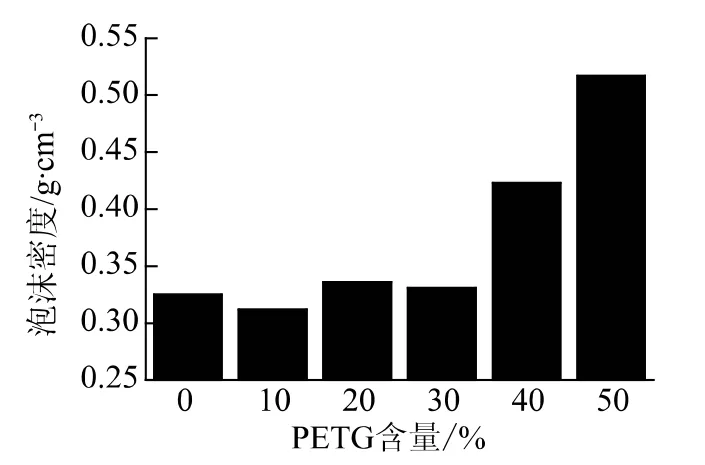
图8 PS和不同PETG 含量PS/PETG 共混物泡沫的泡沫密度Fig.8 Foam density of PS and PS/PETG blends foam with different PETG content
3 结论
(1)在PS中加入PETG 使PS的低频储能模量有一定程度的提高,对应的tanδ也有相应的变化趋势,说明PS/PETG 共混物的熔体弹性要高于纯PS;
(2)PETG 作为分散相,在PS/PETG 共混物中能够起到显著的异相成核作用;
(3)PETG 含量过高时,由于其分散相数量减小,尺寸增大,会导致相应平均泡孔尺寸增大和泡孔密度减小。
[1] Jin W,Xingguo C,Mingjun Y,et al.An Investigationon the Microcellular Structure of Polystyrene/LCP Blends Prepared by Using Supercritical Carbon Dioxide[J].Polymer,2001,42(19):8265-8275.
[2] Zhai W T,Wang H,Yu J,et al.Foaming Behavior of Polypropylene/Polystyrene Blends Enhanced by Improved Interfacial Compatibility[J].Journal of Polymer Science Part B:Polymer Physics,2008,46(16):1641-1651.
[3] Huang H X,Xu H F.Preparation of Microcellular Polypropylene/Polystyrene Blend Foams with Tunable Cell Structure[J].Polymers for Advanced Technologies,2011,22(6):822-829.
[4] 刘本刚,汪文昭,杜中杰,等.不同PETG 对PS/PETG共混物相态结构的影响[J].中国塑料,2013,(11):59-65.Liu Bengang,Wang Wenzhao,Du Zhongjie,et al.Effect of Different PETG on the Morphology Structure of PS/PETG Blends[J].China Plastics,2013,(11):59-65.
[5] Hagui H E,Park C B.Strategies for Achieving Ultra Low-density Polypropylene Foams[J].Polymer Engi-neering and Science,2002,42(7):1481-1492.
[6] Souza A M C,Demarquette.Influence of Composition on the Linear Viscoelastic Behavior and Morphology of PP/HDPE Blends[J].Polymer,2002,43(4):1313-1321.
[7] Márcio Yee,Patrícia S.Calvão,et al.Rheological Behavior of Poly(methyl methacrylate)/Polystyrene(PMMA/PS)Blends with the Addition of PMMA-ran-PS[J].Rheologica Acta,2007,46(5):653-664.
[8] Calvão P S,Yee M,Demarquette N R.Effect of Composition on the Linear Viscoelastic Behavior of PMMA/PS and PMMA/PP Blends[J].Polymer,2005,46(8):2610-2620.
[9] Colton J S,Suh N P.Nucleation of Microcellular Foam:Theory and Practice[J].Polymer Engineering &Science,1987,27:500-503
[10] Colton J S,Suh N P.The Nucleation of Microcellular Thermoplastic Foam with Additives:Part I:Theoretical Considerations [J].Polymer Engineering & Science,1987,27:485-492
[11] Colton J S,Suh N P.The Nucleation of Microcellular Thermoplastic Foam with Additives:Part II:Experimental Results and Discussion[J].Polymer Engineering&Science,1987,27:493-499
[12] Uhlmann D R,Chalmers B,Jackson K A.Interaction Between Particles and a Solid-liquid Interface[J].Journal of Applied Physics,1964,35:2986.
