泰比培南侧链合成工艺改进
2014-03-27周江峰李宏利钏永明季四平袁明龙
周江峰,李宏利,钏永明,季四平,袁明龙,蒋 琳
(云南民族大学 云南省生物高分子功能材料工程技术研究中心,云南 昆明 650500)
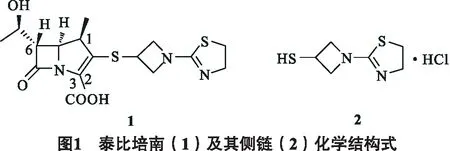
碳青霉烯类抗生素(carbapenem)是20世纪70年代初发展起来的一类全新结构的β-内酰胺酶抑制剂[1-2]. 泰比培南(tebipenem)(1),中文化学名为(1R, 5S, 6S)-6-[(1R)-羟乙基]-1-甲基-2-[1-(1-(2-噻唑啉-2-基)氮杂环丁烷-3-基硫代]-1-碳青霉-2-烯-3-羧酸(见图1),其特戊酯型前药是惠氏制药公司研制的第一个口服碳青霉烯类抗生素,于2009年在日本上市. 泰比培南在临床上多用于儿科呼吸道、泌尿道细菌感染的治疗,对革兰阳性、阴性菌及厌氧菌都具有很强的抗菌活性[3].
泰比培南化学结构具有培南类药物分子的共同特征,即培南母核和侧链通过硫醚键连接,因此泰比培南半合成工艺研究主要是对其侧链合成工艺的改进[4]. 泰比培南侧链2(见图1),中文化学名为1-(4, 5-二氢-2-噻唑啉基)-3-巯基氮杂环丁烷盐酸盐,结构特点为氮杂环丁烷的1位和3位分别被4,5-二氢-2-噻唑啉基和巯基取代.
本实验以苄胺为起始原料,经与环氧氯丙烷取代开环,N-烷基化反应合成1-苄基-3-羟基氮杂环丁烷4;化合物4经氢化脱苄基、N-叔丁氧羰基(Boc)化制备1-Boc-3-羟基氮杂环丁烷5;再经甲磺酰化、脱Boc、连续取代反应、酯交换、成盐6步反应合成泰比培南侧链2,总收率24.3%. 产品结构经1H NMR、HRMS及元素分析确证,泰比培南侧链合成路线见图2.

1 实验部分
1.1 仪器与试剂
Optimelt全自动熔点仪;Varain 400 MHz核磁共振谱仪;Finnigan LCQDECA型液质联用仪;Yanako CHN-Corder元素分析仪.
乙腈使用前用氢化钙回流除水,二甲基亚砜使用前减压蒸馏纯化,其余化学试剂及溶剂均为市售分析纯.
1.2 实验方法
1.2.1 1-苄基-3-羟基氮杂环丁烷(4)的合成
反应烧瓶中加入苄胺 (72.0 g, 670 mmol)和600 mL水,冰浴下滴加环氧氯丙烷(59.0 g, 640 mmol),滴毕反应24 h,滤集析出的白色沉淀,真空干燥得白色固体198 g,为中间体3,77% yield; m.p. 70~72 ℃(lit. 72~73 ℃);中间体3(70.0 g, 350 mmol) 和碳酸氢钠 (59.1 g, 700 mmol) 中加入 560 mL 乙腈,回流24 h,冷却至室温,滤除不溶物,剩余物经石油醚/乙酸乙酯重结晶得白色固体247.2 g(4),82% yield; m.p. 64~65 ℃(lit. 64~65 ℃[5]).
1.2.2 1-叔丁氧羰基-3-羟基氮杂环丁烷(5)的合成
化合物4(40.0 g, 240 mmol) 溶于120 mL甲醇和40 mL水,加入甲酸铵 (30.3 g, 480 mmol) 和钯碳 (10%含量, 10.0 g),室温反应12 h,过滤,滤液于冰浴下加入 (Boc)2O (52.4 g, 240 mmol) 和4-二甲氨基吡啶 (DMAP) (1.5 g, 12 mmol),搅拌8 h,乙酸乙酯提取,有机相减压浓缩,所得残余物经石油醚/乙酸乙酯重结晶得白色固体35.3 g,为中间体5(85% yield);1H NMR (400 MHz, CDCl3)δ:4.53 (m, 1 H), 4.11~4.07 (dd,J= 10.4, 7.2 Hz, 2H), 3.78~3.74 (dd,J=8.8, 4.4 Hz, 2 H), 3.60 (s, 1 H), 1.38 (s, 9 H).
1.2.3 1-叔丁氧羰基-3-甲烷磺酸酯基氮杂环丁烷(6)的合成
化合物5(1.0 g, 5.8 mmol) 溶于20 mL二氯甲烷,加入三乙胺 (1.2 mL, 8.7 mmol),冰浴下滴加甲烷磺酰氯 (0.58 mL, 7.5 mmol),0 ℃反应2 h,反应液依次用水和饱和食盐水洗涤,Na2SO4干燥有机层,减压浓缩溶剂得淡黄色液体粗品1.4 g,为中间体6(96% yield);1H NMR (400 MHz, CDCl3)δ:5.19 (m, 1H), 4.26 (dd,J= 10.0 Hz, 6.8 Hz, 2H), 4.08 (dd,J= 10.0 Hz, 3.6 Hz, 2H), 3.05 (s, 3H), 1.43 (s, 9H).
1.2.4 1-( 4, 5-二氢-2-噻唑啉基)-3-甲烷磺酸酯基氮杂环丁烷(8)的合成
化合物6(1.4 g, 5.6 mmol) 溶于10 mL二氯甲烷,冰浴下加入1 mL三氟醋酸,反应2 h,抽滤得白色固体7(1.31 g, 88% yield);中间体7(0.92 g, 4.9 mmol) 溶于10 mL乙腈,加入2-甲硫基-2-噻唑啉(0.65 g, 4.9 mmol),50 ℃反应6 h,减压浓缩溶剂,残余物溶于水中,饱和碳酸钾水溶液调节pH至9,乙酸乙酯提取,提取液用饱和食盐水洗涤,Na2SO4干燥,浓缩溶剂得白色固体8(0.87 g, 75% yield);1H NMR (400 MHz, CDCl3)δ:5.30 (m, 1H), 4.34 (dd,J= 9.6 Hz, 6.8 Hz, 2H), 4.17 (dd,J= 10.8 Hz, 4.4 Hz, 2H), 4.03 (t,J= 7.6 Hz, 2H), 3.39 (t,J= 7.6 Hz, 2H), 3.07 (s, 3H).
1.2.5 1-( 4, 5-二氢-2-噻唑啉基)-3-硫代乙酸酯基氮杂环丁烷(9)的合成
化合物8(1.0 g, 4.2 mmol) 与硫代乙酸钾 (0.97 g, 8.4 mmol) 溶于10 mL二甲基亚砜(DMSO),110 ℃反应3 h,乙酸乙酯稀释,依次用水、饱和食盐水洗涤,Na2SO4干燥,减压浓缩溶剂,残余物经快速柱层析纯化,V(石油醚)∶V(丙酮)=2∶1得黄色液体0.76 g,为中间体9(yield 84%);1H NMR (400 MHz, CDCl3)δ:4.43 (t,J= 8.4 Hz, 2H), 4.30 (m, 1H), 4.01 (t,J=7.6 Hz, 2H), 3.89 (dd,J= 9.2 Hz, 5.6 Hz, 2H), 3.36 (t,J= 7.6 Hz, 2H), 2.34 (s, 3H).
1.2.6 1-( 4, 5-二氢-2-噻唑啉基)-3-巯基氮杂环丁烷盐酸盐(2)的合成
化合物9(500 mg, 2.3 mmol) 溶于甲醇 (10 mL),加入甲醇钠 (128.8 mg, 2.3 mmol),室温下搅拌0.5 h,加入浓度为2 mol/L的氯化氢甲醇溶液2.8 mL (5.5 mmol),反应15 min后过滤除去不溶物,滤液减压浓缩,乙腈/四氢呋喃重结晶得无色晶体,为目标产物2(413 mg, 85% yield); m.p. 132~134 ℃, (lit. 134~136 ℃[5]);1H NMR (400 MHz, CDCl3)δ:12.19 (s, 1H), 5.26~5.19 (m, 1H), 4.63 (t,J= 7.6 Hz, 2H), 4.18~4.02 (m, 4H), 3.59 (t,J= 7.6 Hz, 2H), 2.57 (d,J= 7.2 Hz, 1H); HRMS (FAB) calcd for C6H11N2S2175.0364, found 175.0376 (M-Cl); Anal. Calcd for C6H11ClN2S2: C, 34.19; H, 5.26; N, 13.29, found: C, 34.17; H, 5.18; N, 13.24.
2 结果与讨论
已知文献报道的泰比培南侧链合成路径主要分为3类:①以二苯甲胺为起始原料,首先合成中间体3-羟基氮杂环丁烷盐酸盐10,后对其氨基和羟基进行转化,完成4,5-二氢-2-噻唑啉基和巯基的引入(图3a)[6];②以烯丙基胺为起始原料,在合成氮杂环丁烷片段的同时构建3-巯基片段,得到中间体3-巯基氮杂环丁烷盐酸盐11后再引入二氢噻唑片段(图3b)[7-8];③从苄胺出发,合成N-苄基-3-羟基氮杂环丁烷,经甲烷磺酰化、氢化脱苄基2步操作得3-甲烷磺酸酯基氮杂环丁烷盐酸盐7,随后再完成二氢噻唑和巯基片段的构建(图3c)[5,9].
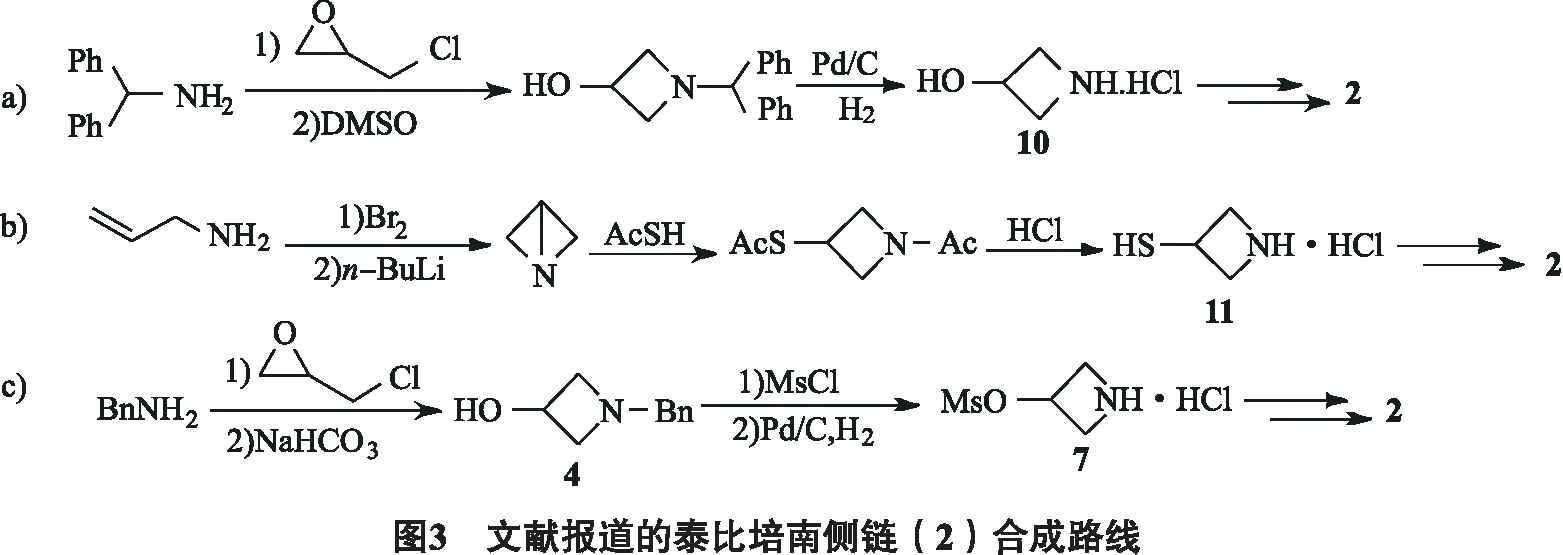
1)与上述合成路径a, b相比,改进后的合成路线避免了强吸湿,大极性中间体10或11的合成或直接分离纯化;由于路径c中用氢气/钯碳脱苄基时产物7在反应条件下易发生开环,新方法在较早阶段即对氮上保护基进行更换,使用在常规酸碱条件下即可快速引入及脱除的Boc基团替换苄基,化合物4脱苄后可在反应体系中原位与(Boc)2O反应而无需对脱苄产物进行分离. 通过筛选氢源,确定了以甲酸铵作为氢源在甲醇中反应,脱苄产物能够很好地耐受该反应条件,此外,中间体5无需经柱层析或是重结晶纯化,粗品即可用于后续投料.
2)改进的合成路线中,中间体6~8粗品即可满足后续反应需求,如需进一步制得纯品可通过重结晶操作,无需柱层析纯化.
3)改进的合成路线中,中间体6用三氟醋酸脱Boc反应收率高,产物7性质稳定,未观察到类似路径c中氢化条件下的开环副反应.
3 结语
本法从廉价易得的苄胺出发,经10步反应合成碳青霉烯类抗生素泰比培南侧链,总收率24.3%,产品结构经1H NMR、HRMS及元素分析确证. 改进后的合成路线革除了文献报道方法中毒性大、成本高的反应试剂,反应条件温和,操作简便,收率稳定,具有较好的放大生产前景.
参考文献:
[1] 邓玲华, 李虹影. 碳青霉烯类抗生素的特点与应用[J]. 中国全科医学, 2004, 7(2): 104-105.
[2] 董耘婷, 张永信. 碳青霉烯类抗生素的发展与展望[J]. 上海医药, 2011, 32(7): 316-319.
[3] 张文君, 吴文芳, 冯小龙. 新型口服碳青霉烯类抗菌药物——泰吡培南酯[J]. 河北医药, 2010, 32(18): 2596-2599.
[4] 黄小光, 陈矛, 朱少璇, 等. 替比培南匹伏酯及其侧链的合成研究进展[J]. 化学与生物工程, 2011, 28(12): 20-23.
[5] ISODA T, YAMAMURA I, TAMAI S, et al. A practical and facile synthesis of azetidine derivatives for oral carbapenem, L-084[J]. Chem Pharm Bull, 2006, 54(10): 1408-1411.
[6] 黄小光, 黄冰娥, 朱少璇. 替比培南匹伏酯侧链的合成[J]. 中国新药杂志, 2012, 21(4): 428-430.
[7] HAYASHI K, SATO C, HIKI S. Novel efficient synthesis of 1-azabicyclo[1.1.0]butane and its application to the synthesis of 1-(1,3-thiozolin-2-yl)azetidine-3-thiol useful for the pendant moiety of an oral 1(-methylcarbapenem antibiotic L-084[J]. Tetrahedron Lett,1999, 40(19): 3761-3764.
[8] HAYASHHI K, HIKI S, KUMAGAI T. Synthesis of azetidine derivatives using 1-azabicyclo[1.1.0] butane[J]. Heterocycles, 2002, 56(1/2): 433-442.
[9] ISODA T, TAMAI S, KUMAGAI T. An efficient method for the synthesis of 3-mercapto-1-(1,3-thiazolin-2-yl)azetidine useful for the pendant moiety of oral carbapenem, L-084[J]. Heterocycles,2006, 68(9):1821-1824.
