Differential microRNA expression between shoots and rhizomes in Oryza longistaminata using high-throughput RNA sequencing
2014-03-13YingZongLiyuHungTingZhngQioQinWenshengWngXiuqinZhoFengyiHuBinyingFuZhikngLi
Ying Zong,Liyu Hung,Ting Zhng,Qio Qin,Wensheng Wng,Xiuqin Zho,Fengyi Hu,Binying Fu,*,Zhikng Li
aInstitute of Crop Science/National Key Facility for Crop Gene Resources and Genetic Improvement,Chinese Academy of Agricultural Sciences,Beijing 100081,China
bFood Crops Research Institute,Yunnan Academy of Agricultural Sciences,Kunming 650205,China
1.Introduction
Rice is a staple food for more than half of the world's population,and rice cultivation is the largest single food-producing use of land,covering 9% of the Earth's arable land [1].Growing annual rice on steep hillsides causes soil erosion,reducing farm productivity and damaging resources downhill.Breeding perennial rice varieties with rhizomes is an effective way to solve this problem.Annual soil disturbances associated with tillage would be avoided through the use of a perennial cultivar,and rhizomes would trap soil,preventing erosion.Among the two cultivated and 22 wild rice species studied,Oryza longistaminata is a wild,perennial species from Africa that is characterized by the presence of rhizomatous stems[2,3].Rhizomes enable O.longistaminata to overwinter,producing new plants in the following growing season.O.longistaminata is the only perennial rice species with the AA genome,allowing it to be used as a donor in breeding programs for perennial upland rice [4,5].However,partial-sterility barriers have impeded the development of perennial rice by conventional breeding [6].
Genetic studies show that the rhizomatous trait in rice is quantitatively controlled by many genes.In our previous study,an F2and two backcross populations from O.longistaminata and RD23 were used for genetic mapping of the rhizomatous trait.The results revealed two dominant complementary genes that controlled rhizome expression:Rhz2 and Rhz3,which mapped to rice chromosomes 3 and 4,respectively[7].A comparative gene expression analysis between aerial shoots (ASs) and rhizomes was performed to identify organ-specific gene expression,and the results indicated that 2566 genes,including transcription factors,were differentially expressed in ASs and rhizomes.A few of these genes were found colocalized in the rhizome-related QTL intervals[8].Further profiling revealed that primary metabolites and hormones had distinct organ distribution patterns.Metabolites accumulated in stem bases and a higher indole-3-acetic acid-to-zeatin riboside ratio is probably associated with the regulatory metabolism of rhizome formation [9].These data suggest that rhizome development in O.longistaminata is controlled by a complex molecular genetic network.
Recently,microRNAs(miRNAs)have emerged as regulators of many key biological functions in plants including the processes of organogenesis and morphogenesis.In Arabidopsis,genetic deficiencies associated with miRNAs can cause the plant to grow abnormally.For example,a mutation in the triplet of miR164 can cause a severe disruption of shoot development[10].miR824 plays an important role in stomatal complex formation in Arabidopsis [11,12].In tomato,the miR393 target gene LA influences compound leaf development via a miRNA binding site mutation[13].Several miRNAs have been identified in rice,including those associated with root growth [14],grain development[15,16],seed development[17],leaf morphogenesis and growth [18,19],and plant architecture [20].Whether miRNAs are involved in the molecular regulation of rhizome development in O.longistaminata is still unknown.
In this study,high-throughput RNA sequencing was performed to profile miRNA expression in the ASs and rhizomes of O.longistaminata.The comprehensive miRNA expression data,with their tissue-specific expression patterns,provide further information on the functional genomics of O.longistaminata as well as molecular evidence for elucidating the regulatory mechanisms of rhizome development.
2.Materials and methods
2.1.Plant materials and growth conditions
The wild rice O.longistaminata,with long and strong rhizomes,was used in this study.It was originally collected in Niger and cultured in the greenhouse at the Institute of Crop Science,Chinese Academy of Agricultural Sciences (Beijing,China;latitude 39.9°N,longitude 116.3°E).Two tissues:ASs,including stem tips,the topmost internodes and the youngest leaf,and rhizomes,including rhizome tips and internodes,were collected at the active tillering stage and flash frozen in liquid nitrogen.
2.2.Small RNA library construction and sequencing
Total RNA was extracted from sampled AS or rhizome tissues of the three biological replicates using the TRIzol reagent(Invitrogen,USA).The quality and concentration of the RNA were evaluated by spectrophotometer and gel electrophoresis.Small-RNA sequencing was performed by CapitalBio Corporation,Beijing,China.Two small RNA libraries for the ASs and rhizomes were constructed using TruSeq Small RNA Sample Prep Kit(Illumina)according to the manufacturer's protocol.Briefly,4 μg of total RNA was ligated to the 3′-adapter and the 5′-adapter.Single-stranded cDNA was synthesized by reverse transcription(RT).Then 140 to 160 bp fragments were selected by gel purification to produce small RNA libraries for cluster generation and sequencing.The primary data analysis and base calling were performed using the Illumina instrument's software.
2.3.Sequencing data analysis of the small RNAs
Individual sequence reads with base quality scores were produced by Illumina.Adaptor and low-quality sequences were removed,and all identical sequences were counted and eliminated from the initial data set.The unique reads were mapped to the rice genome of the Rice Genome Annotation Project(RGAP)at Michigan State University (MSU) [21] using the program Bowtie[22].Sequences that matched rice rRNA,scRNA,snoRNA,snRNA,or tRNA sequences in the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) and Rfam RNA family databases were filtered out [23,24].In addition,sequences shorter than 17 nt or longer than 35 nt and those overlapping exons and introns in the mRNAs,were also removed.Sequences that perfectly matched miRNA precursors and mature miRNAs in the Sanger miRBase(http://www.mirbase.org/,release 20 June 2013) of rice were identified as known miRNAs.The sequences that matched miRBase entries of other plant species,but not rice,were designated as conserved miRNAs.To identify potentially novel miRNAs,the software Mireap(http://sourceforge.net/projects/mireap/)was used to predict precursor sequences and their secondary structures.
To obtain potential gene targets for the identified miRNAs,the online tools psRNA target (http://plantgrn.noble.org/psRNATarget/) [25] and WMD3 (http://wmd3.weigelworld.org/cgibin/webapp.cgi) [26] were used to query rice cDNAs of RGAP at MSU2 (http://rice.plantbiology.msu.edu/) that had scores of less than 3.
A web tool,IDEG6 [27],was employed to identify differentially expressed miRNAs in ASs and rhizomes.The expression of miRNAs in the two tissues was normalized to transcripts per million(TPM),and then miRNAs with P values lower than 0.001 and fold changes of greater than 2.0 or lower than 0.5 were identified as significantly differently expressed between the two tissues.
2.4.Expression confirmation of miRNA by quantitative RT-PCR
Total RNA was isolated from ASs and rhizomes of O.longistaminata using TRIzol reagent.DNA contamination was removed by incubating with RNase-free DNase I(NEB,USA)for 45 min at 37 °C.Approximately 2 μg of total RNA was reversetranscribed in a 20 μL reaction volume using the miRcute miRNA cDNA Synthesis Kit(TIANGEN,China).The tailing reactions were incubated for 60 min at 37 °C,followed by the RT reaction at 37 °C for 60 min.cDNA templates for miRNA targets were synthesized using Oligo dT primers and the Fermentas RevertAid First Strand cDNA Synthesis Kit (Fermentas,USA) according to the manufacturer's instructions.U6 snRNA was chosen as the internal control for miRNA expression and actin as the internal control for miRNA target gene expression.The expression levels of the miRNAs and the corresponding target genes were validated through the ABI Step One Plus Real-Time PCR System (Applied Biosystems,USA) using the SYBR Premix Ex Taq kit (Takara,Japan).The miRNA cDNAs were diluted 4 times,and 2 μL of diluted product was mixed with 10 μL of 2*SYBR reaction mix and 0.2 μL (200 nmol L-1final concentration) of each of the miRNAspecific forward and universal reverse primers in a 20 μL PCR amplification mixture.The cDNAs for the target genes were diluted 20 times.Two-step PCR reactions were performed with the following cycling parameters: 30 s at 95 °C,followed by 35 cycles of 10 s at 95 °C and 31 s at 57 °C.The results were represented as the mean ± SD of the three replicates.A melting curve analysis was carried out for each PCR product to identify nonspecific amplification[28,29].All primers for real-time RT-PCR are listed in Tables S1 and S2.
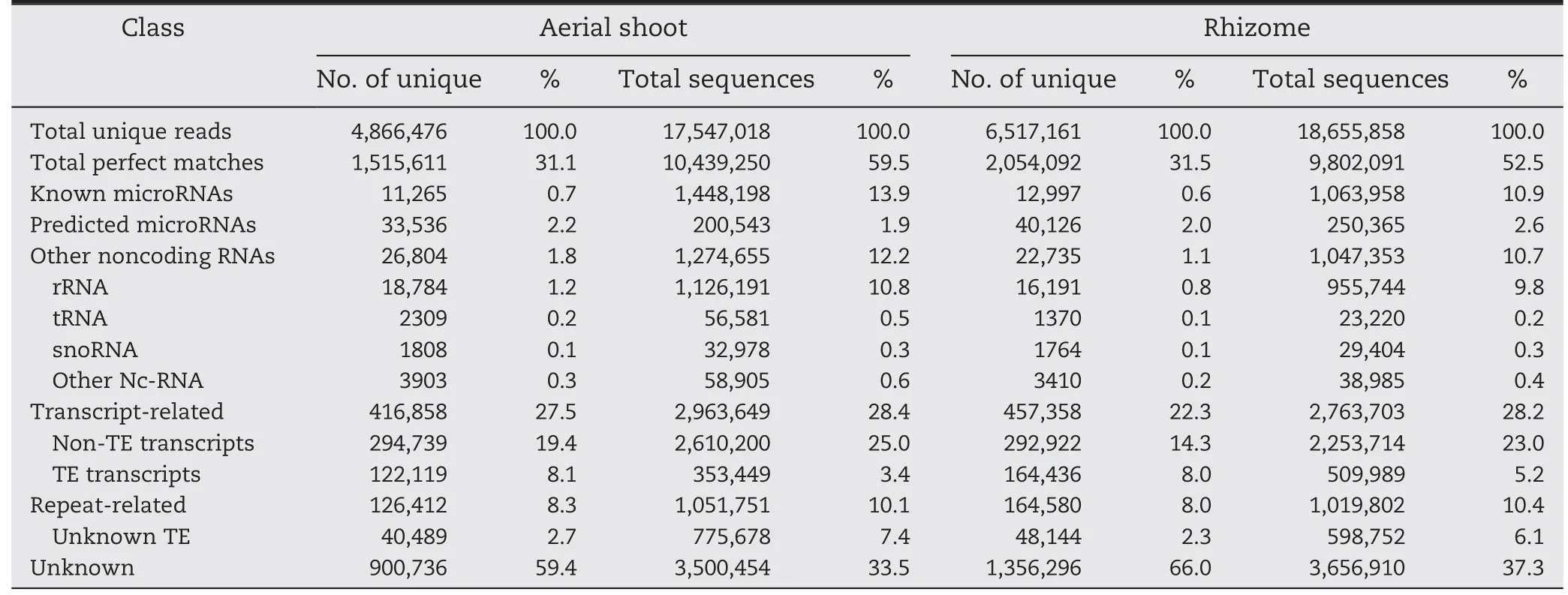
Table 1-Distribution of small RNAs among different categories in aerial shoots and rhizomes of Oryza longistaminata.
2.5.Target gene validation by RNA ligase-mediated 5′amplification of cDNA ends(RLM-RACE)
To determine the miRNA cleavage sites in the target genes,RLM-RACE was performed using the SMARTer RACE cDNA Amplification Kit (Clontech,PT4096-2).First,total RNA was extracted from the two tissues and ligated with SMARTer II A oligonucleotide,and then the RNA was reverse transcribed using 10 × Universal Primer A Mix (UPM).PCR was then performed twice,using the UPM/gene-specific primer in the first reaction and the UPM/nested gene-specific primer in the second,according to the manufacturer's instructions.The product was then gel-extracted and cloned into the PMD20-T Vector(Takara,Dalian,China)for sequencing.The primers for RLM-RACE are shown in Table S3.
3.Results
3.1.High-throughput sequencing of small RNAs in O.longistaminata
To investigate the small RNA expression profiles of O.longistaminata,two cDNA libraries of small RNAs,one from ASs and one from rhizomes,were sequenced.In total,20,358,337 raw reads from ASs and 21,313,971 from rhizomes were produced.After elimination of low quality reads,adaptors and contaminating sequences,17,547,018 and 18,655,858 clean reads with lengths of 17 to 30 nt remained from the ASs and rhizomes,respectively.Of these reads,4,866,476 and 6,517,161,respectively,were unique (Table 1).The overall size distributions of the sequenced reads from the two libraries were very similar,with the 24 nt class being the most abundant(Fig.1).
The unique sequences were mapped to the rice genome assembly using Bowtie [22].As shown in Table 1,almost every category of RNAs,including miRNAs,siRNAs,rRNAs,snoRNAs,snRNAs,tRNAs,repeat-associated sRNAs,and degraded mRNAs,were detected in both tissues.Finally,11,265 and 33,536 reads for ASs,and 12,997 and 40,126 reads for rhizomes were identified as known and predicted miRNA candidates,respectively,for analysis.
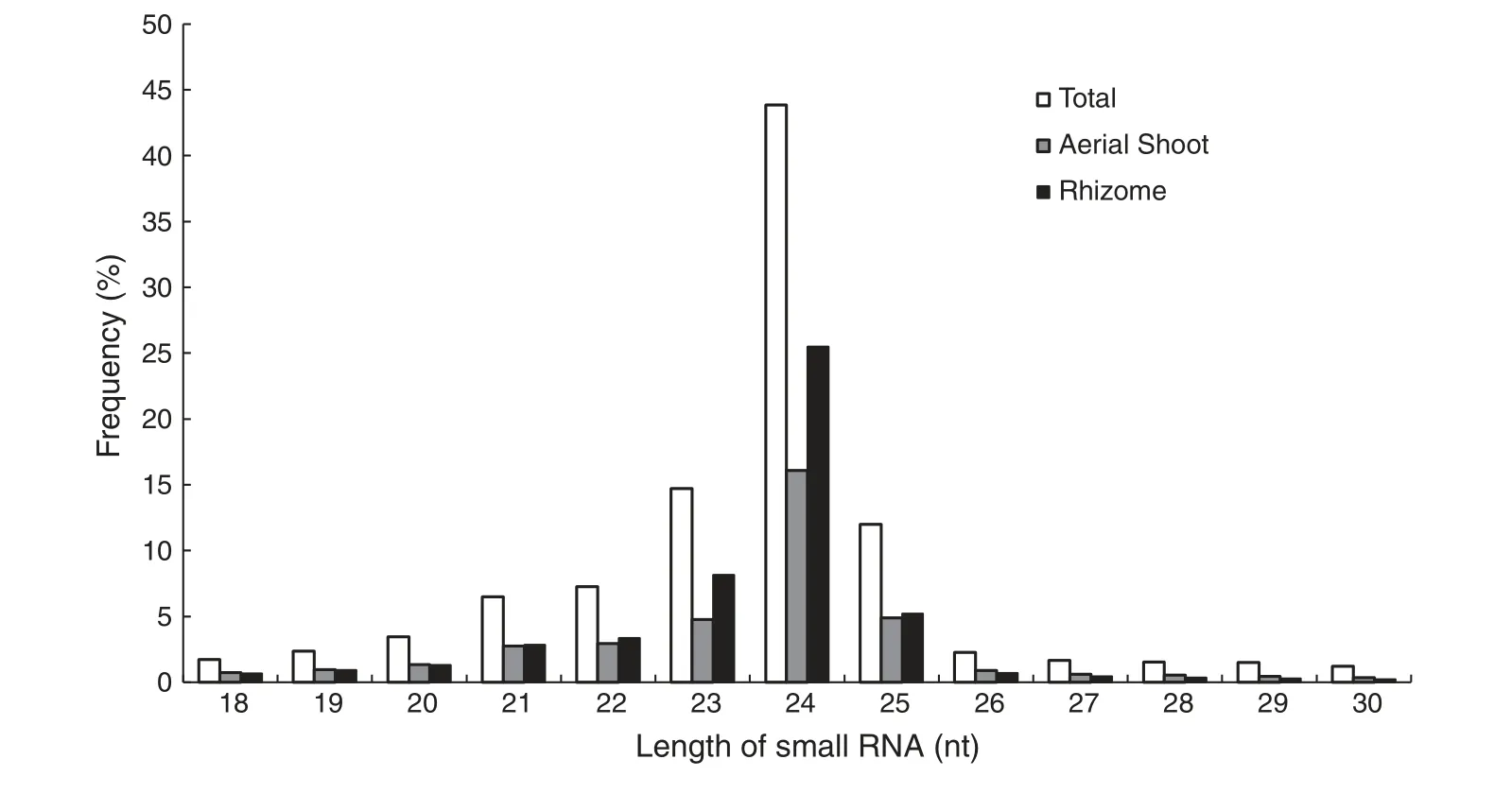
Fig.1-Length distribution of small RNAs from aerial shoots and rhizomes of Oryza longistaminata.Vertical axis represents the frequency of small RNA in different size.Horizontal axis shows small RNA in different size.
3.2.Analysis of differentially expressed known,conserved and novel miRNAs in ASs and rhizomes
All small RNA sequences were searched against the plant miRNA database to identify known,conserved and novel miRNAs in ASs and rhizomes,as described in Materials and methods.To reduce false-positive rates,only sequences with at least two detected reads were designated as miRNA candidates.Of the 713 known rice miRNAs deposited in the miRBase database (Release 20,June 2013),380 known rice miRNAs were identified as being expressed in ASs and rhizomes,including 340 miRNAs found in both tissues(Table 2).Among them,21 and 19 known miRNAs were expressed exclusively in ASs and in rhizomes,respectively(Fig.2,Tables 2,S4).The most highly tissue-specific miRNAs included osa-miRNA319a-3p and osa-miRNA529a in the rhizomes and osa-miRNA530-5p and osa-miRNA5073 in the ASs,indicating their roles in rhizome and AS growth.
In the conserved and novel miRNAs 72 conserved miRNAs were expressed,including 53 miRNAs common to ASs and rhizomes.Seven and 12 miRNAs were expressed specifically in ASs and rhizomes,respectively (Table S5).A total of 151 novel miRNAs were identified in both tissues,including nine and 19 miRNAs expressed exclusively in ASs and rhizomes,respectively (Table S6).These results indicated that some miRNAs were expressed in a tissue-specific manner,implying their roles in specific tissue development.
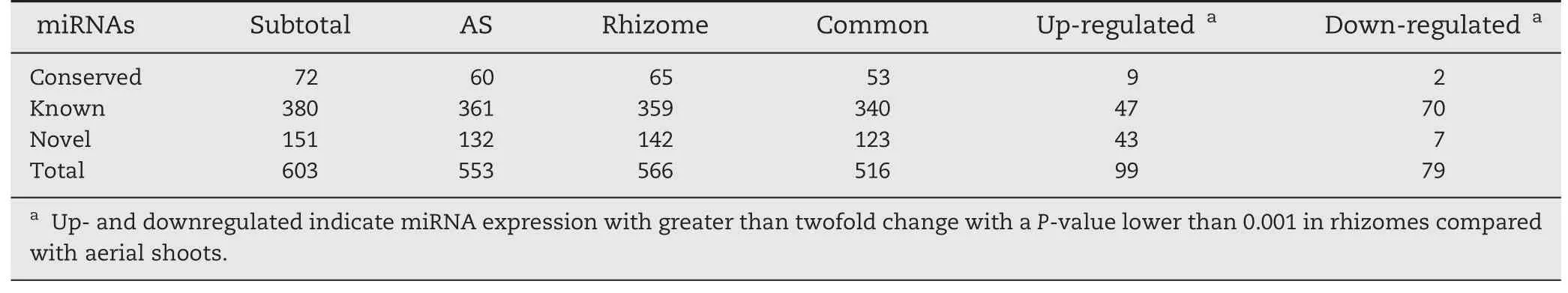
Table 2-Expression profiling of known,conserved,and novel miRNAs in aerial shoots and rhizomes of Oryza longistaminata.
The expression patterns of the identified miRNAs in the ASs and rhizomes were comparatively analyzed to identify differentially expressed miRNAs.Overall,178 miRNAs were differentially expressed with a greater than twofold change and a P-value lower than 0.001 in ASs and rhizomes(Table 2).These included 47 and 70 known miRNAs whose expression levels were up-or downregulated,respectively,in rhizomes compared with ASs(Tables 2,S4).Interestingly,several miRNA family members,including 10 members of osa-miR156,four of osa-miR164,three of osa-miR393,16 of miR395,and seven of osa-miR444,were found simultaneously downregulated in rhizomes relative to ASs.Additionally,three members of osamiR169,seven of osa-miR1861,three of osa-miR2118,three of osa-miR5148,five of osa-miR819,and three of miR812 were upregulated in rhizomes compared with ASs(Table 3).
The expressions of eight miRNAs detected to be expressed in the AS and rhizome were confirmed by qRT-PCR.Results showed that four miRNAs: osa-miR156a,osa-miR159a.1,osamiR393,and osa-miR444b.2,were identified as highly enriched in ASs compared with rhizomes.This result was consistent with the sequencing results that indicated lower expression levels in rhizomes by 0.44,0.49,0.22,and 0.39 fold changes,respectively,compared with ASs.The other miRNAs,including osa-miR160d and novel-17b*,were also confirmed to be differentially expressed in the ASs and rhizomes by qRT-PCR(Fig.S1).
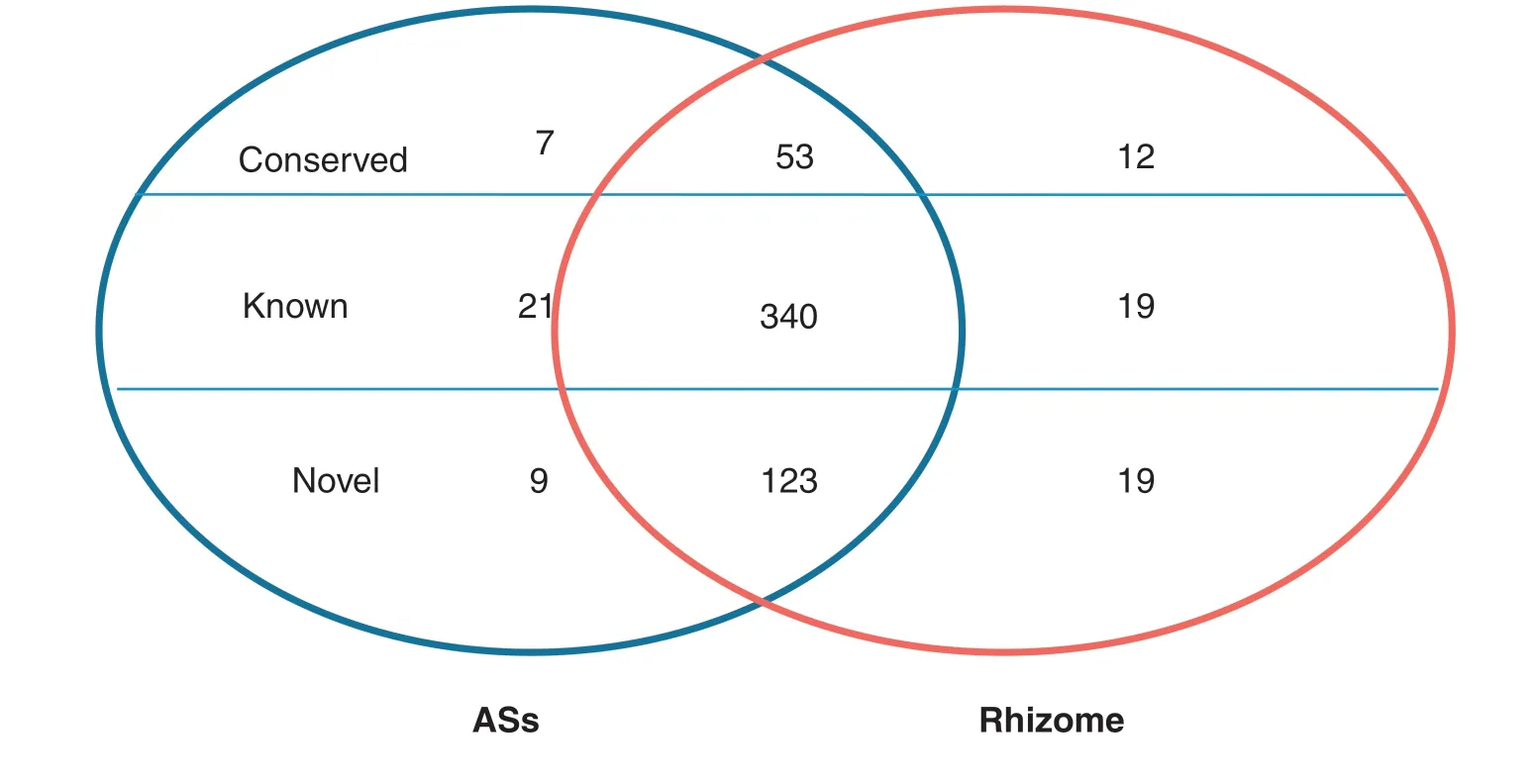
Fig.2-Known,conserved,and novel microRNAs in aerial shoots and rhizomes of Oryza longistaminata represented in a Venn diagram.The two ellipses represent aerial shoots (AS)and rhizomes.Each ellipse consists of three parts,from top to bottom:conserved,known and novel microRNAs.Overlapping areas represent microRNAs common to both tissues.
3.3.Target prediction and identification of miRNAs exclusively or differentially expressed in rhizomes
To better understand the biological roles of miRNAs in the ASs and rhizomes of O.longistaminata,the putative target genes for the detected miRNAs were identified as described in the Materials and Methods.In total,2996 potential target genes for 290 miRNAs were identified,with an average of 10.33 targets per miRNA.Table S7 shows the 144 predicted targets of the miRNAs expressed exclusively or differentially in rhizomes compared with ASs,including 17 known rice miRNAs or miRNA families,seven newly identified novel miRNAs,and two conserved miRNAs.A total of 62 of the 144 target genes were transcription factors,including 19 MADSboxes,17 SBPs,10 nuclear transcription factors,four ARFs,two TCPs,and two ERFs.Other target genes included those involved in signal transduction,metabolism,stress response,and programmed cell death.Gene Ontology analysis of these 144 targets indicated that these genes were highly involved in transcription regulation,metabolic processes,cellular processes,and reproduction (Fig.S2).
An RLM-RACE experiment was performed to verify that the miRNAs could induce the cleavage of the corresponding target(s).Sequence analysis of the 5′RACE cleaved products showed that a predicted target gene of miRNA156a was squamosa promoterbinding-like protein 10(SPL10),and the cleavage site mediated by osa-miR156a was detected in the transcript of SPL10 (LOC_Os02g07780.1).The relationships of miRNAs and their targets were also verified for osa-miR156a and two genes encoding teosinte glume architecture 1 (TGA1) (Fig.S3).The MADS-box transcription 23 gene for osa-miR444b.2,a TCP TF gene (LOC_Os07g05720.1) for osa-miR319b,an expressed protein gene(LOC_Os09g36650.1) for osa-miR159a.1,a gene encoding a putative protein(LOC_Os04g07260.1)for osa-miR319a-5p,and a gene encoding biopterin transport-related BT1(LOC_Os03g58080.1)for osa-miR5148a were also confirmed,as shown in Fig.S2.It was noted that cleavage might occur upstream or downstream of the binding site instead of the commonly observed position.For example,the binding site of LOC_Os08g33488.1 (target of osamiR444b.2) occurred between 311 and 331 bp; however,the cleavage site occurred at about 360 bp,downstream of the binding site,which is consistent with previous reports[30,31].
Quantitative RT-PCR was performed to determine the expression relationship between miRNAs and their corresponding targets,as shown in Fig.S4 and Table S4.In contrast to the lower expression of osa-miR156a in the rhizome,the expression levels of its targets,two TGA1s,were highly enriched in the rhizome compared with the AS.However,the expression of another target in the two tissues,SPL10,was similar to those of osa-miR156a.The transcripts of osa-miR319b and its target gene TCP were simultaneously identified as highly enriched in rhizome compared with AS(Figs.S2,S4).These results indicated that miRNAs could be negatively or positively involved in the regulation of their targets at the post-transcriptional level.
4.Discussion
The development of high-throughput gene expression analyses,including deep sequencing techniques,has enabled the rapid profiling and investigation of the transcriptome.In O.longistaminata,genome-wide gene expression profiling has previously been performed using the Affymetrix rice microarray to identify tissue-specific genes,in particular genes related to rhizome development [8].In this study,the comparative analysis of two small RNA libraries,one from ASs and one from rhizomes,indicated that some miRNAs were differentially expressed in the two tissues,and target gene predictions for these differentially expressed miRNAs suggested their roles in AS and rhizome development.
MiRNAs play an important role in plant growth and development.To date,there are 592 miRNA sequences representing 713 mature miRNAs in the rice miRBase(http://www.mirbase.org/cgi-bin/mirna_summary.pl?org = osa).However,the miRNA transcriptome of wild rice,including O.longistaminata,is poorly characterized[32].In the present study,380 known rice miRNAs were identified in ASs and rhizomes,indicating that the majority of the identified rice miRNAs could be expressed in O.longistaminata.In addition,72 conserved miRNAs and 151
putative novel miRNAs were found to be expressed in the ASs and rhizomes of O.longistaminata,providing a rich resource for the further elucidation of small RNA functions in rice.
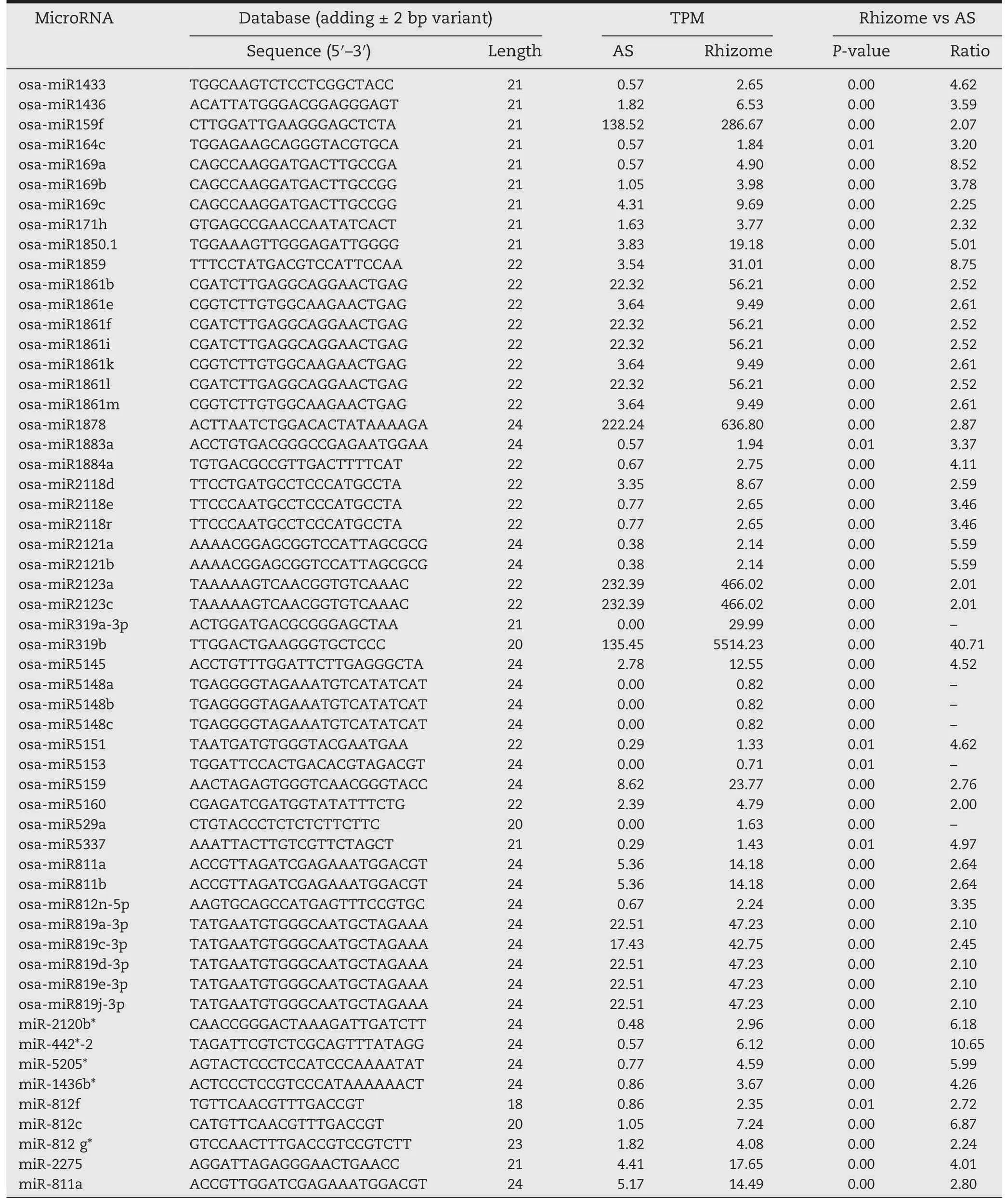
Table 3-Highly enriched known and conserved miRNAs in rhizomes compared with aerial shoots of Oryza longistaminata.
Many miRNAs display temporal or tissue-specific expression patterns [33].Some miRNAs were expressed exclusively in ASs and rhizomes of O.longistaminata,indicating their possible regulatory roles in tissue development.We identified 19 miRNAs,including osa-miR319a-3p and osa-miR529a,which were highly and exclusively expressed in the rhizome,and four predicted target genes for osa-miR319a-3p were characterized as encoding the Alg9-like mannosyltransferase protein,dihydrodipicolinate reductase,LSD ONE LIKE 3(LOL3),and a retrotransposon protein(Table S4).LOL3 is a zinc finger that may be involved in programmed cell death and defense responses[34].While the targets for osa-miR529a were predicted to encode a carboxyl-terminal proteinase,a phytosulfokine receptor,a conserved hypothetical protein,and a transposon protein (Table S4),their detailed functions in rhizome development need further investigation.
Comparative analysis of miRNAs differentially expressed between ASs and rhizomes could promote understanding of miRNA functions in rhizome growth regulation and development.In this study,117 known rice miRNAs,including several important miRNA families,were found to be differentially expressed in rhizomes relative to ASs.Ten members of the osa-miR156 family,whose target genes are TGA1,SBP TFs,and SPL TFs,which were previously reported to be related to growth and development in plants [35–37],had significantly lower expression levels in rhizomes than in ASs.Seven members of the osa-miR444 family,whose predicted target genes included several MADS-box TFs and SNF2 TF,which were found to be involved in cellular processes,also had lower expression levels in rhizomes [38,39].In contrast,osa-miR319b,whose target genes are two TCP TFs,which have been reported to control the morphology of shoot lateral organs[40],was highly enriched in the rhizome.These results revealed that the identified differentially expressed miRNAs,correlated with their respective target genes,could function in the regulation of rhizome formation.
miRNAs bind to target sequences in mRNAs,typically resulting in repressed gene expression,and targets can also reciprocally control the level and function of miRNAs[41].In the present study,expression antagonism was observed for several miRNAs and their corresponding target genes,including osa-miR156a and two TGA1s.However,a correlative antagonistic expression pattern could not be detected for osa-miR319b and its target TCP gene,indicating their co-expression in specific tissues,a finding consistent with previous reports[42,43].These results imply the existence of other mechanisms,such as feedback regulation between miRNAs and their targets[15,44,45],in addition to the reciprocal regulatory mechanism.
In conclusion,a genomewide miRNA expression analysis from ASs and rhizomes of O.longistaminata was performed using high-throughput small RNA sequencing.A set of miRNAs was determined to be exclusively or differentially expressed in the two tissues.The results of target gene predictions suggest that the differentially expressed miRNAs are involved in the regulatory control of tissue development,especially rhizome formation,in a complex way.
Supplementary material related to this article can be found http://dx.doi.org/10.1016/j.cj.2014.03.005.
Acknowledgment
This work was supported by the National Natural Science Foundation of China(31271694 and U1302264).
[1] FAO,WFP and IFAD,The State of Food Insecurity in the World.Economic growth is necessary but not sufficient to accelerate reduction of hunger and malnutrition,FAO,Rome,2012.
[2] A.Ghesquiere,Re-examination of genetic control of the reproductive barrier between Oryza longistaminata and O.sativa and relationship with rhizome expression,Rice Genetics II,International Rice Research Institute,The Philippines,1991.729–730.
[3] D.A.Vaughan,The Wild Relatives of Rice: A Genetic Resources Handbook,IRRI,The Philippines,1994.46–47.
[4] D.Tao,F.Hu,Y.Yang,P.Xu,J.Li,G.Wen,Rhizomatous individual was obtained from interspecific BC2F1progenies between Oryza sativa and Oryza longistaminata,Rice Genet.Newsl.18(2001) 11–13.
[5] E.J.Sacks,M.P.Dhanapala,D.Y.Tao,Breeding for perennial growth and fertility in an Oryza sativa/O.longistaminata population,Field Crops Res.95(2006) 39–48.
[6] E.J.Sacks,V.Schmit,K.L.McNally,Fertility in an interspecific rice population and its effect on selection for rhizome length,Field Crops Res.95(2006) 30–38.
[7] F.Y.Hu,D.Y.Tao,E.Sacks,B.Y.Fu,P.Xu,J.Li,Y.Yang,K.McNally,G.S.Khush,A.H.Paterson,Z.K.Li,Covergent evolution of perenniality in rice and sorghum,Proc.Natl.Acad.Sci.U.S.A.100 (2003) 4050–4054.
[8] F.Y.Hu,D.Wang,X.Q.Zhao,T.Zhang,H.Sun,L.H.Zhu,F.Zhang,L.Li,Q.Li,D.Y.Tao,B.Y.Fu,Z.K.Li,Identification of rhizome-specific genes by genome-wide differential expression Analysis in Oryza longistaminata,BMC Plant Biol.11(2011)18.
[9] X.Q.Zhao,T.Zhang,L.Y.Huang,H.M.Wu,F.Y.Hu,F.Zhang,L.H.Zhu,B.Y.Fu,Comparative metabolite profiling and hormone analysis of perennial and annual Rice,J.Plant Biol.55(2012) 73–80.
[10] P.Sieber,F.Wellmer,J.Gheyselinck,J.L.Riechmann,E.M.Meyerowitz,Redundancy and specialization among plant microRNAs:role of the MIR164 family in developmental robustness,Development 134 (2007) 1051–1060.
[11] E.R.Alvarez-Buylla,S.J.Liljegren,S.Pelaz,S.E.Gold,C.Burgeff,G.S.Ditta,F.Vergara-Silva,M.F.Yanofsky,MADS-box gene evolution beyond flowers:expression in pollen,endosperm,guard cells,roots and trichomes,Plant J.24(2000)457–466.
[12] C.Kutter,H.Schöb,M.Stadler,F.Meins,A.Si-Ammour,MicroRNA-mediated regulation of stomatal development in Arabidopsis,Plant Cell 19(2007) 2417–2429.
[13] N.Ori,A.R.Cohen,A.Etzioni,A.Brand,O.Yanai,S.Shleizer,N.Menda,Z.Amsellem,I.Efroni,I.Pekker,Regulation of LANCEOLATE by MIR319 is required for compound-leaf development in tomato,Nat.Genet.39 (2007) 941–951.
[14] X.Ma,C.Shao,H.Wang,Y.Jin,Y.Meng,Construction of small RNA-mediated gene regulatory networks in the roots of rice(Oryza sativa),BMC Genomics 14(2013) 510.
[15] R.Yi,Z.Zhu,J.Hu,Q.Qian,J.Dai,Y.Ding,Identification and expression analysis of microRNAs at the grain filling stage in rice (Oryza sativa L.) via deep sequencing,PLoS ONE 8(3)(2013) e57863.
[16] Y.C.Zhang,Y.Yu,C.Y.Wang,Z.Y.Li,Q.Liu,J.Xu,J.Y.Liao,X.J.Wang,L.H.Qu,F.Chen,P.Xin,C.Yan,J.Chu,H.Q.Li,Y.Q.Chen,Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching,Nat.Biotechnol.31 (2013) 848–852.
[17] K.A.Watanabe,P.Ringler,L.Gu,Q.J.Shen,RNA-sequencing reveals previously unannotated protein-and microRNA-coding genes expressed in aleurone cells of rice seeds,Genomics 103 (2013) 122–134.
[18] K.Xie,J.Shen,X.Hou,J.Yao,X.Li,J.Xiao,L.Xiong,Gradual increase of miR156 regulates temporal expression changes of numerous genes during leaf development in rice,Plant Physiol.158 (2012) 1382–1394.
[19] C.Yang,D.Li,D.Mao,X.Liu,C.Ji,X.Li,X.Zhao,Z.Cheng,C.Chen,L.Zhu,Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice(Oryza sativa L.),Plant Cell Environ.36(2013) 2207–2218.
[20] Y.Jiao,Y.Wang,D.Xue,J.Wang,M.Yan,G.Liu,G.Dong,D.Zeng,Z.Lu,X.Zhu,Q.Qian,J.Li,Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice,Nat.Genet.42 (2010) 541–544.
[21] S.Ouyang,W.Zhu,J.Hamilton,H.Lin,M.Campbell,K.Childs,F.Thibaud-Nissen,R.L.Malek,Y.Lee,L.Zheng,J.Orvis,B.Haas,J.Wortman,C.R.Buell,The TIGR rice genome annotation resource: improvements and new features,Nucl.Acids Res.35(Suppl.1)(2007) D883–D887.
[22] B.Langmead,C.Trapnell,M.Pop,S.L.Salzberg,Ultrafast and memory-efficient alignment of short DNA sequences to the human genome,Genome Biol.10 (3)(2009) R25.
[23] P.P.Gardner,J.Daub,J.G.Tate,E.P.Nawrocki,D.L.Kolbe,S.Lindgreen,A.C.Wilkinson,R.D.Finn,S.Griffiths-Jones,S.R.Eddy,A.Bateman,Rfam: updates to the RNA families database,Nucl.Acids Res.37 (2009) 136–140.
[24] T.Barrett,S.E.Wilhite,P.Ledoux,C.Evangelista,I.F.Kim,M.Tomashevsky,K.A.Marshall,K.H.Phillippy,P.M.Sherman,M.Holko,A.Yefanov,H.Lee,N.Zhang,C.L.Robertson,N.Serova,S.Davis,A.Soboleva,NCBI GEO:archive for functional genomics data sets–update,Nucl.Acids Res.41(D1)(2013)D991–D995.
[25] X.Dai,P.X.Zhao,PsRNATARGET:a plant small RNA target analysis server,Nucl.Acids Res.39 (2011) 155–159.
[26] S.Ossowski,R.Schwab,D.Weigel,Gene silencing in plants using artificial microRNAs and other small RNAs,Plant J.53(2008) 674–690.
[27] C.Romualdi,S.Bortoluzzi,F.D'Alessi,G.A.Danieli,IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments,Physiol.Genomics 12(2006) 159–162.
[28] J.M.Franco-Zorrilla,F.J.Del Toro,M.Godoy,J.Pérez-Pérez,I.López-Vidriero,J.C.Oliveros,G.García-Casado,C.Llave,R.Solano,Genome-wide identification of small RNA targets based on target enrichment and microarray hybridizations,Plant J.59(2009) 840–850.
[29] Z.Xu,S.Zhong,X.Li,W.Li,S.J.Rothstein,S.Zhang,Y.Bi,C.Xie,Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots,PLoS ONE 6(11) (2011) e28009.
[30] R.Chen,Z.Hu,H.Zhang,Identification of microRNAs in wild soybean(Glycine soja),J.Integr.Plant Biol.51(2009)1071–1079.
[31] C.Zeng,W.Wang,Y.Zheng,X.Chen,W.Bo,S.Song,W.Zhang,M.Peng,Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants,Nucl.Acids Res.38(2010) 981–995.
[32] Y.Wang,X.Bai,C.Yan,Y.Gui,X.Wei,Q.H.Zhu,L.Guo,L.Fan,Genomic dissection of small RNAs in wild rice (Oryza rufipogon): lessons for rice domestication,New Phytol.196(2012) 914–925.
[33] D.Mittal,S.K.Mukherjee,M.Vasudevan,N.S.Mishra,Identification of tissue-preferential expression patterns of rice miRNAs,J.Cell.Biochem.114 (2013) 2071–2081.
[34] P.Epple,A.A.Mack,V.R.Morris,J.L.Dangl,Antagonistic control of oxidative stress-induced cell death in Arabidopsis by two related,plant-specific zinc finger proteins,Proc.Natl.Acad.Sci.U.S.A.100 (2003) 6831–6836.
[35] J.C.Preston,L.C.Hileman,Functional evolution in the plant SQUAMOSA-promoter binding protein-like(SPL)gene family,Front.Plant Sci.4(2013) 80.
[36] J.C.Preston,H.Wang,L.Kursel,J.Doebley,E.A.Kellogg,The role of teosinte glume architecture (tga1) in coordinated regulation and evolution of grass glumes and inflorescence axes,New Phytol.193 (2012) 204–215.
[37] L.Z.Ling,S.D.Zhang,Exploring the evolutionary differences of SBP-box genes targeted by miR156 and miR529 in plants,Genetica 140 (2012) 317–324.
[38] J.A.Eisen,K.S.Sweder,P.C.Hanawalt,Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions,Nucl.Acids Res.23(1995) 2715–2723.
[39] M.M.Kater,L.Dreni,L.Colombo,Functional conservation of MADS-box factors controlling floral organ identity in rice and Arabidopsis,J.Exp.Bot.57(2006) 3433–3444.
[40] T.Koyama,M.Furutani,M.Tasaka,M.Ohme-Takagi,TCP transcription factors control the morphology of shoot lateral organs via negative regulation of the expression of boundary-specific genes in Arabidopsis,Plant Cell 19(2007)473–484.
[41] A.E.Pasquinelli,MicroRNAs and their targets: recognition,regulation and an emerging reciprocal relationship,Nat.Rev.Genet.13(2012) 271–282.
[42] H.Liu,I.S.Kohane,Tissue and process specific microRNA–mRNA co-expression in mammalian development and malignancy,PLoS ONE 4(2009) e5436.
[43] F.Wang,L.Li,L.Liu,H.Li,Y.Zhang,Y.Yao,Z.Ni,J.Gao,High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage(Brassica rapa L.ssp.pekinensis),Mol.Genet.Genomics 287 (2012) 555–563.
[44] D.Baulcombe,RNA silencing in plants,Nature 431 (2004)356–363.
[45] L.J.Xue,J.J.Zhang,H.W.Xue,Characterization and expression profiles of miRNAs in rice seeds,Nucl.Acids Res.37(2009) 916–930.
杂志排行

The Crop Journal的其它文章
- Effect of nitrogen fertilizer on distribution of starch granules in different regions of wheat endosperm
- Induced defense responses in rice plants against small brown planthopper infestation
- Overexpression of GmDREB1 improves salt tolerance in transgenic wheat and leaf protein response to high salinity
- Impacts of nighttime post-anthesis warming on rice productivity and grain quality in East China
- Establishment of the integrated applied core collection and its comparison with mini core collection in soybean(Glycine max)
- Genome-wide analysis of the WRKY transcription factor gene family in Gossypium raimondii and the expression of orthologs in cultivated tetraploid cotton