Genome-wide analysis of the WRKY transcription factor gene family in Gossypium raimondii and the expression of orthologs in cultivated tetraploid cotton
2014-03-13CaipingCaiErliNiuHaoDuLiangZhaoYueFengWangzhenGuo
Caiping Cai,Erli Niu,Hao Du,Liang Zhao,Yue Feng,Wangzhen Guo*
State Key Laboratory of Crop Genetics & Germplasm Enhancement,Hybrid Cotton R&D Engineering Research Center,MOE,Nanjing Agricultural University,Nanjing 210095,China
1.Introduction
Transcription factors,which exist in all living organisms,are essential for the regulation of gene expression.WRKY transcription factors,a family of regulatory genes,were first identified in plants[1–3].In WRKY family proteins,a 60 amino acid region is highly conserved among family members.It includes the conserved WRKYGQK sequence followed by one of the two types of zinc finger motifs,the C2H2and C2–HC types[4].All known WRKY proteins can be divided into three groups (group I,II,and III) based on the number of WRKY domains and the types of zinc finger motif.Two WRKY domains can be found in group I proteins,whereas a single domain is present in group II and group III proteins.Generally,group I and group II proteins share the same C2H2-type zinc finger motif (C–X4–5–C–X22–23–H–X1–H).In group III,WRKY domains contain a C2–HC-type motif(C–X7–C–X23–H–X1–C)[4].Group II is further classified into several subgroups based on their phylogenetic clades[4–6].
In plants,WRKY proteins form a large family of transcription factors and are known to function in response to various physiological processes.WRKY transcription factors are important components of many aspects in the plant defense system,including MAMP-triggered immunity (MTI) or PAMPtriggered immunity (PTI),effector-triggered immunity (ETI),and systemic acquired resistance [7–14].These transcription factors also play important regulatory roles in plant abiotic stress.For example,Arabidopsis plants that overexpress GmWRKY21 are more cold-stress tolerant than wild-type plants,and plants overexpressing GmWRKY54 exhibit increased salt and drought tolerance,whereas plants overexpressing GmWRKY13 exhibit increased sensitivity to salt and mannitol stress[15].In barley(Hordeum vulgare),HvWRKY38 is involved in cold and drought responses[16].The expression of AtWRKY25 and AtWRKY26 is induced upon treatment with high temperatures,whereas AtWRKY33 expression is repressed in response to the same treatment [17].In addition to functioning in biotic and abiotic stresses,WRKY transcription factors regulate developmental processes,such as trichome and seed coat development in Arabidopsis [18],sesquiterpene biosynthesis in cotton (Gossypium hirsutum)[19],seed development in barley,Solanum chacoense,and Arabidopsis [20–22],and senescence in Arabidopsis[23–25].
Since the release of a large number of publicly available sequences and even complete whole-genome sequences in some plants,genome-wide analyses of the WRKY gene family have been performed.There are at least 72 WRKY family members in Arabidopsis[4],more than 100 in rice(Oryza sativa)[5],57 in Cucumis sativus[26],104 in Populus trichocarpa[27],and 81 in Solanum lycopersicum [28].Genome duplication events have been detected in this family [27],and the divergence of the monocots and dicots was verified based on the analysis of WRKY transcription factors[5,6].
The genus Gossypium has great economic and scientific importance.Cotton produces the most important natural textile fiber in the world and is also an important oilseed crop.Cotton fiber is an outstanding model for studying plant cell elongation and cell wall biosynthesis[29].Tetraploid cotton is also an excellent model system for studying polyploidization and genome duplication.Despite the importance of WRKY genes in plant growth and developmental processes,to our knowledge only eight WRKY genes have previously been reported from different cotton species [13,19,30,31].Genomewide analysis of the WRKY transcription factor family in Gossypium will lay the foundation for elucidating their structure,evolution,and functional roles.
Currently 435,344 cotton EST sequences are available in the GenBank EST database(http://www.ncbi.nlm.nih.gov/dbEST/).Among them,297,214 ESTs were identified in G.hirsutum,63,577 in Gossypium raimondii,41,781 in Gossypium arboreum,32,525 in Gossypium barbadense,and 247 in Gossypium herbaceum.A pilot study for the whole-genome scaffold sequence of the diploid cotton G.raimondii,which is the putative contributor of D-subgenome to fiber-producing cotton species including G.hirsutum and G.barbadense,has been released by two research groups [32,33].As an application,G.raimondii genome sequences have been of great advantage for assembling the tetraploid transcriptome and mining candidate genes of interest [34].Information from the publicly available Gossypium database will serve as a foundation for identifying gene families such as WRKY genes.
The objective of the current study was to survey the WRKY genes and their phylogenetic relationship in Gossypium with a bioinformatic approach using information derived from the publicly available database from the two drafts of the D5genome (G.raimondii) and ESTs from NCBI (http://www.ncbi.nlm.nih.gov/dbEST/),combined with sequence data confirmation via cloning of cDNAs containing complete open reading frames (ORFs) from upland cotton.We further evaluated the expression patterns of WRKY genes in various developmental stages and under various stress conditions in tetraploid cultivated cotton species.
2.Materials and methods
2.1.Prediction of WRKY gene family
Genes and proteins annotated in G.raimondii were downloaded from http://www.phytozome.net/ and http://cgp.genomics.org.cn/.WRKY transcription factors were identified using HMMER software version 3.0[35]and the PFAM protein family database using the WRKY domain (PF03106) as a query [36].Expressed sequence tag (EST) sequences for four cotton species,G.hirsutum (Gh),G.barbadense (Gb),G.arboreum (Ga),and G.raimondii (Gr),were downloaded from the GenBank EST database (http://www.ncbi.nlm.nih.gov/dbEST/).WRKY protein sequences in Arabidopsis were obtained from The Arabidopsis Information Resource(TAIR:http://www.arabidopsis.org/).
2.2.Mapping and analysis of WRKY genes
Mapping of WRKY genes was performed using MapInspect(http://www.plantbreeding.wur.nl/UK/ software_mapinspect.html).Exons and introns were predicted by comparing the coding sequences with their genomic sequences using the online GSDS program [37].Conserved motif prediction was performed using the MEME program [38].The following parameters were used for analysis: maximum number of motifs,10; minimum motif width,six; and maximum motif width,70.
2.3.Sequence alignment and phylogenetic construction
Alignment of the amino acid sequences of the WRKY domain with approximately 60 amino acids was performed with ClustalX 1.83 [39].The parameters used in the alignment were as follows: for pairwise parameters,gap opening: 10.00,gap extension: 0.10,protein weight matrix: Gonnet 250; for multiple parameters,gap opening: 10.00,gap extension: 0.20,delay divergent sequence (%): 30,DNA transition weight: 0.50,use negative matrix:OFF,protein weight matrix:Gonnet series;for protein gap parameters,residue-specific penalties: ON,hydrophilic penalties: ON,hydrophilic residues: GPSNDQEKR,gap separation distance:0,end gap separation:ON.A maximum likelihood tree was used to construct the phylogenetic tree based on the bootstrap method (number of bootstrap replications: 1000) and the Poisson model using MEGA 5.0 software[40].
2.4.Plant materials and treatments
G.hirsutum accession TM-1,a genetic standard line of upland cotton,was used for tissue/organ expression analysis.The plants were grown in the field under normal conditions.Petals and anthers were sampled on the day of flowering,and ovules and fibers were excised from developing flower buds or bolls on selected days post anthesis(DPA).Roots,stems,and leaves were collected from two-week-old seedlings.All tissues collected were quick-frozen in liquid nitrogen and stored at-70 °C before use.
G.hirsutum cultivar Jinmian 19,which exhibits high tolerance to abiotic stress,was used for the abiotic stress treatments.Salt and drought stress treatments were applied by immersing the seedlings in 200 mmol L-1NaCl and 20%PEG-6000,respectively.The leaves were harvested at appropriate times,quick-frozen in liquid nitrogen,and stored at-70 °C before use.
Gossypium barbadense cultivar Hai 7124,which exhibits Verticillium resistance,was used for fungal pathogen(V.dahliae) inoculation.The roots of Hai 7124 seedlings were dipped in V.dahliae strain VD8 conidial suspensions containing 107spores mL-1.The roots were harvested at the appropriate time,quick-frozen in liquid nitrogen,and stored at-70 °C before use.
2.5.RNA isolation and real-time PCR analysis
Total RNA was isolated according to the method of Jiang and Zhang [41].To remove genomic DNA,the RNA samples were treated with DNase I.First-strand cDNA was synthesized based on reverse transcription of 2 μg RNA digested by DNase I using the reverse transcription polymerase reaction system(Promega,USA).For real-time PCR,gene-specific primers were designed based on the WRKY gene sequences using Primer 5.0(http://www.premierbiosoft.com/).The amplified fragment length ranged from 75 bp to 200 bp,and the annealing temperature ranged from 58 °C to 60 °C.The cotton histone3(AF024716) gene (forward primer and reverse primer sequences 5′-GAAGCCTCATCGATACCGTC-3′ and 5′-CTACC ACTACCATCATGG-3′,respectively)was used as the reference gene[19].
The amplification reactions of the real-time PCR were performed using an ABI 7500 real-time PCR system.The amplification parameters were as follows: denaturation at 95 °C for 10 min,40 cycles of denaturation at 95 °C for 15 s,annealing at 58–60 °C for 15 s,and extension at 72 °C for 15 s.For the melting curve stage,the default settings were chosen.Three biological replicates,each with three technical replicates,were tested.The expression levels of the WRKY genes were calculated according to Livak and Schmittgen[42].
2.6.Cloning and sequencing of WRKY cDNAs in upland cotton
Based on bioinformatic analysis,gene-specific PCR primer pairs were individually designed for PCR-amplification of the WRKY genes based on the complete ORF cDNA sequences(Table S1),and the transcripts from various tissues of G.hirsutum acc.TM-1 were used for amplification.
Standard PCR analysis was performed using High-Fidelity ExTaq DNA Polymerase [TaKaRa Biotechnology (Dalian) Co.,Ltd.,China].The PCR products were cloned into the pMD18-T vector (TaKaRa) according to the manufacturer's instructions and sequenced from plasmid DNA templates.At least six randomly selected clones for each gene were subjected to sequencing.The cDNA sequences of the WRKY genes were determined using alignment analysis with their corresponding sequences obtained from bioinformatic analysis.
3.Results
3.1.Genome-wide exploration of WRKY genes and their chromosome distribution
Both the whole genome sequence scaffolds of two drafts of the D5genome [32,33] and ESTs from four cotton species(http://www.ncbi.nlm.nih.gov/) were used for genome-wide exploration of WRKY genes in genus Gossypium.Using HMMER software version 3.0 [35] and the PFAM protein family database with the WRKY domain(PF03106)[36],we identified a total of 120 WRKY transcription factors based on the sequence information from Paterson et al.[32].Of these transcription factors,103 homologous WRKY genes were also found based on the sequence information of Wang et al.[33].However,there were differences in the lengths of the proposed sequences of 33 WRKY genes,ranging from 3 bp to 1797 bp,as determined by performing sequences comparison between the two D5genome databases (Table S2).These differences may have been due largely to assembly error in partial chromosomal regions and require further confirmation.Furthermore,3668 ESTs,including 519 from G.raimondii,2935 from G.hirsutum,148 from G.barbadense,and 70 from G.arboreum,were found to match these WRKY members with at least one EST hit (e ≤-10).When the WRKY genes were compared with the sequences in the Arabidopsis database from TAIR (http://www.arabidopsis.org/),105 WRKY homologs in Arabidopsis were also detected with BLASTn (e ≤-10)analysis(Table S2).Integrating the above results,we identified a total of 120 candidate WRKY genes in G.raimondii with corresponding expressed sequence tags found in at least one of four cotton species,including tetraploid cultivated cotton species G.hirsutum and G.barbadense,diploid cultivated cotton species G.arboreum and G.raimondii.
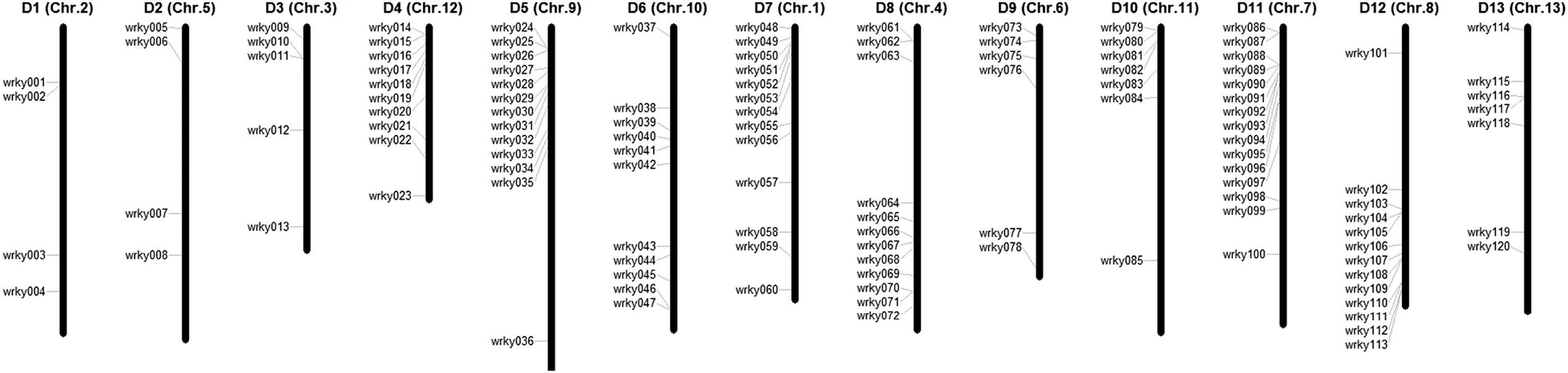
Fig.1-Reordering of 13 scaffolds of G.raimondii and chromosome distribution of WRKY genes.The consensus map was constructed based on the collinearity of the newly updated interspecific genetic map in allotetraploid cultivated cotton species reported recently [43] and the 13 chromosomes of the G.raimondii genome [32].The chromosome numbers from D1 to D13 refer to the map of Zhao et al.[43],and the names of the 13 scaffolds from the G.raimondii genome are shown in parentheses.The size of each chromosome is indicated based on its relative length.The candidate WRKY genes are designated WRKY1 to WRKY120 following their orders on chromosomes.

Fig.2-Phylogenetic tree of WRKY domains in Gossypium and Arabidopsis.The phylogenetic tree was constructed with MEGA 5.0 software using the maximum-likelihood method based on the 60 conserved amino acid WRKY domains.The WRKY gene with suffix“N”or “C” indicated the NTWD or CTWD.
To characterize the chromosomal distribution of these WRKY genes,we integrated 13 scaffolds of the G.raimondii genome(named Chr.1 to Chr.13)from Paterson et al.[32]with a previously reported high-density interspecific genetic map of allotetraploid cultivated cotton species[43].The collinearity between the genetic map and the cotton D5genome revealed homologs between 13 Dt chromosomes in tetraploid cotton species and 13 scaffolds of G.raimondii.We reordered the 13 scaffolds of G.raimondii according to the corresponding D1 to D13 chromosomes in tetraploid cotton species [43].As a result,120 candidate WRKY genes were matched to 13 scaffolds of the D5genome and were designated WRKY1 to WRKY120 based on the order of the homologs on chromosomes D1 to D13.The distribution of WRKY family members on the 13 chromosomes was uneven,with the fewest (four)members located on D1 and on D2 and the most(15)members located on D11(Fig.1).
3.2.Classification,structure,and variation of WRKY genes in Gossypium
Of the 120 candidate cotton WRKY genes,we detected 140 WRKY domain regions spanning approximately 60 amino acids,with 18 WRKY candidates containing two WRKY domains and one(WRKY108)containing three WRKY domains(Fig.S1).By comparison of the amino acid sequences in the WRKY domain regions from Gossypium and Arabidopsis,120 cotton WRKY candidate genes were classified into three groups (groups I,II,and III),and group II genes were further classified into five subgroups (groups IIa–e; Fig.2),based on the classification rules employed for the WRKY family genes in Arabidopsis[4].
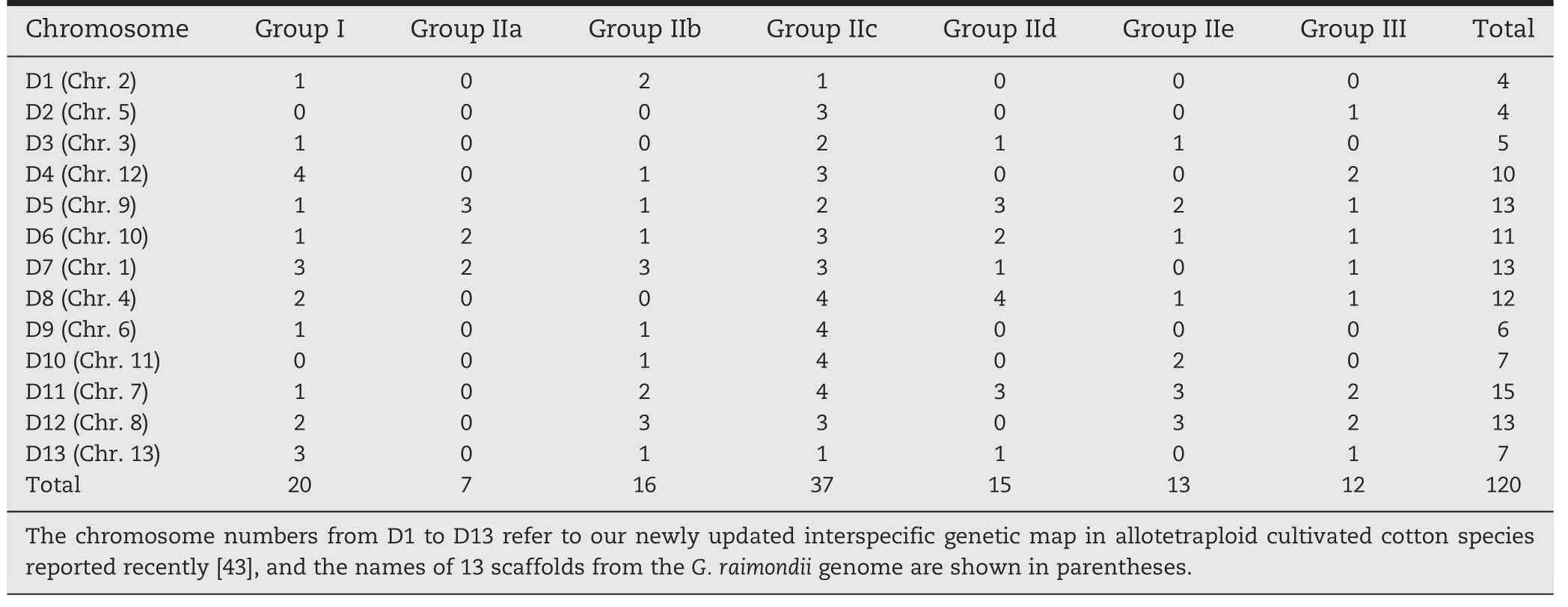
Table 1-Classification and genome distribution of WRKY transcription factor family genes.
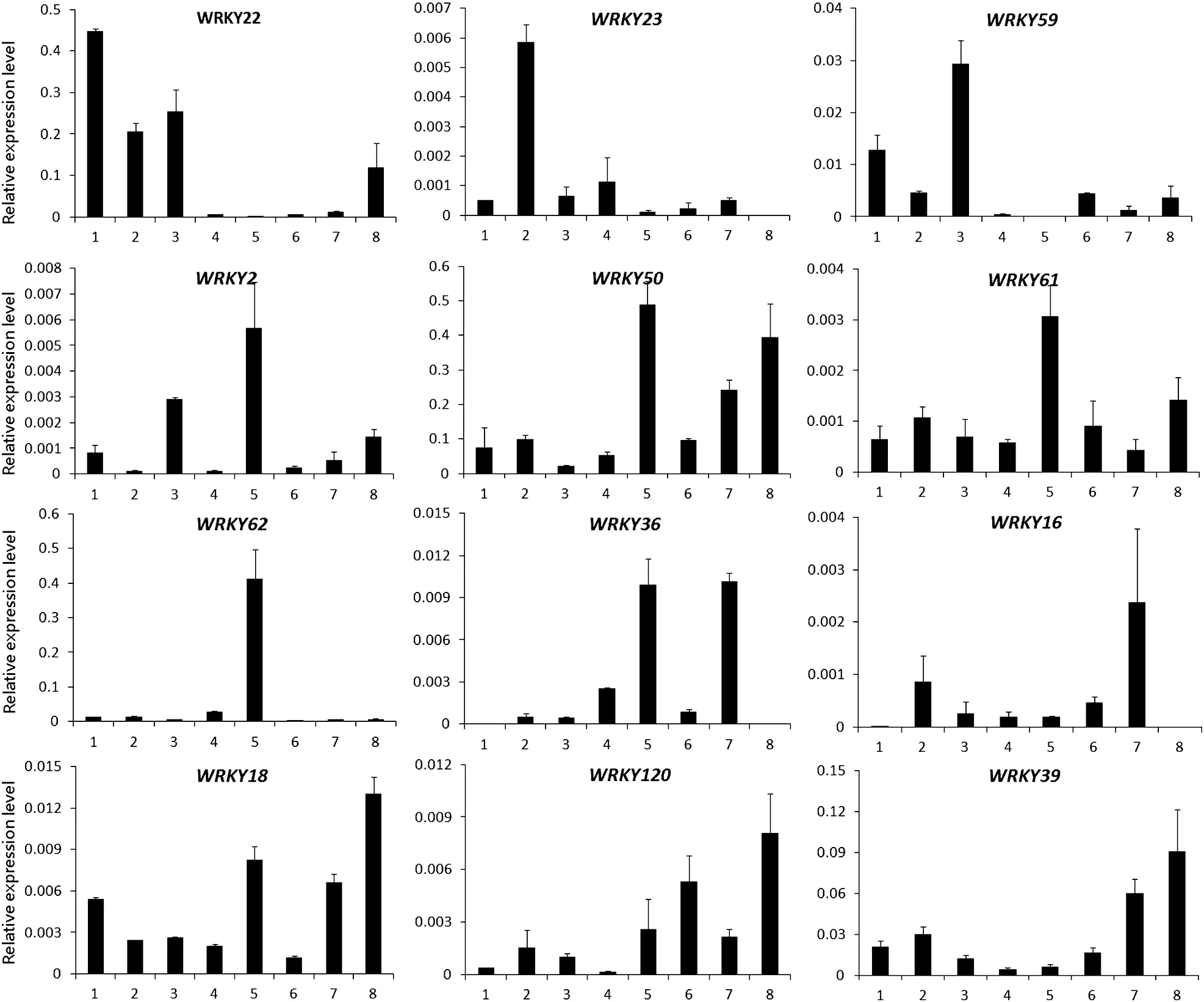
Fig.3-Expression patterns of WRKY genes from group I in various tissues by qRT-PCR.The Y-axis indicates relative expression levels and the X-axis indicates different tissues.1:root;2:stem;3:leaf;4:petal;5:anther;6:0 PDA;7:10 PDA;8:21 PDA.The error bars were calculated based on three biological replicates using standard deviation.The cotton histone3(AF024716)gene was used as the reference gene.
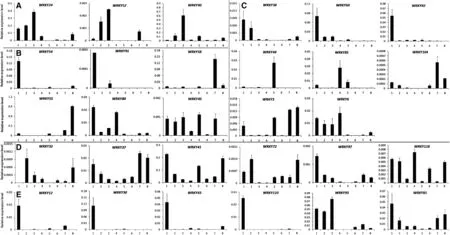
Fig.4-Expression patterns of WRKY genes from group II in various tissues by qRT-PCR.A: Expression patterns of WRKY genes from group IIa; B: Expression patterns of WRKY genes from group IIb; C: Expression patterns of WRKY genes from group IIc; D: Expression patterns of WRKY genes from group IId; E: Expression patterns of WRKY genes from group IIe.1: root; 2: stem; 3: leaf; 4: petal; 5: anther; 6: 0 PDA; 7: 10 PDA; 8: 21 PDA.
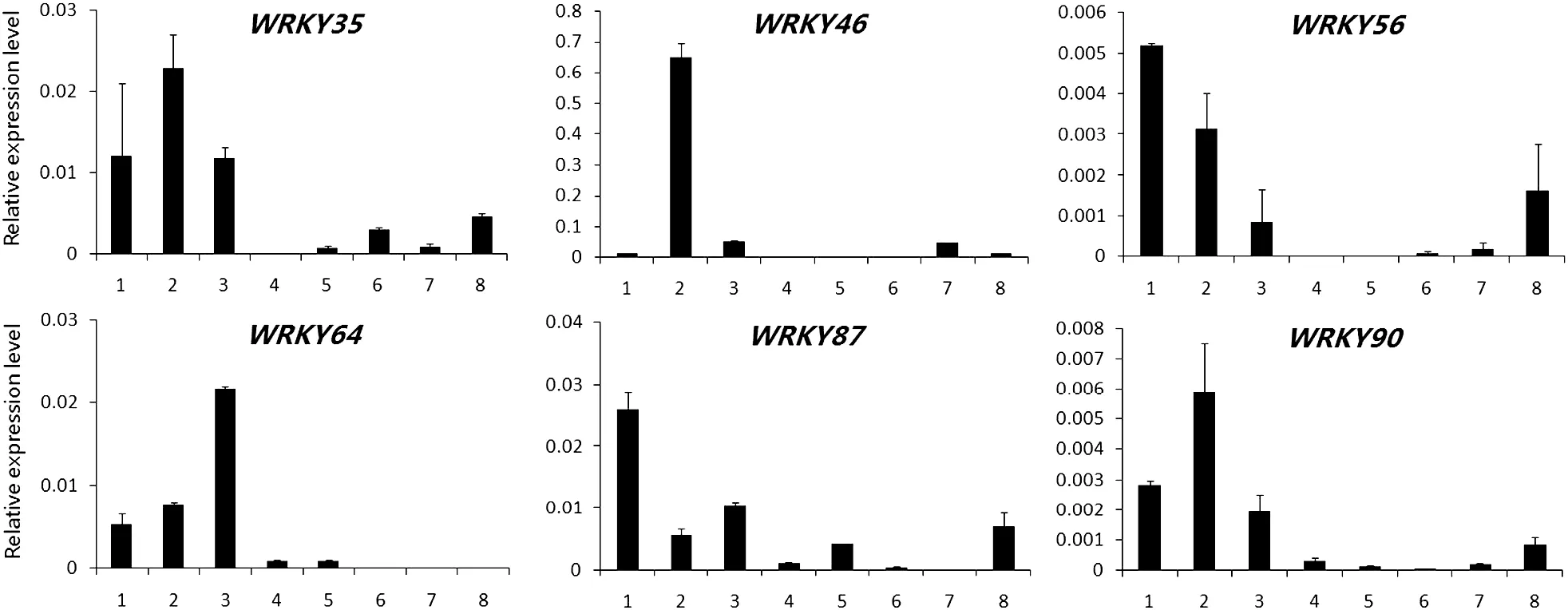
Fig.5-Expression patterns of WRKY genes from group III in various tissues by qRT-PCR.1:root;2:stem;3:leaf;4:petal;5:anther;6:0 PDA;7:10 PDA;8:21 PDA.
Among the three groups,there were 20 members in group I,88 in group II,and 12 in group III.Furthermore,in group II,subgroups IIa–e contained 7,16,37,15,and 13 members,respectively.The types and chromosome distribution of these members are described in Table 1.It is noteworthy that WRKY108 in group I contained three WRKY domains(WRKY108N1,WRKY108N2,and WRKY108C).However,the three WRKY domains were not clustered in the N-terminal WRKY domain (NTWD) and the C-terminal WRKY domain(CTWD).The phylogenetic results showed that WRKY108N1,WRKY108N2,and WRKY108C were clustered into group IIc,group III,and group IId,respectively(Fig.2).
According to D5genomic sequence information,there was at least one intron insert in the WRKY candidate genes,with WRKY108 and WRKY109 having the most complex structures.The intron splices in the conserved WRKY domain could be classified into two major types,the R type and the V type.V-type introns were observed only in groups IIa and IIb (Fig.S2).In addition to the WRKY domain,the WRKY family members were also predicted by MEME to contain other conserved motifs.However,six WRKY proteins,encoded by WRKY14,WRKY21,WRKY35,WRKY46,WRKY77,and WRKY90,contained only a WRKY domain(Fig.S3).
WRKYGQK residues are considered to be important regions of the WRKY transcription factor family.However,we found some genes with diverse amino acid residues in this region.Among the seven amino acid residues(WRKYGQK),mutations at the W and K sites were not observed; most variations involved Q to T,H,or K substitutions.For WRKY109 in group I,there were large variations in this seven residue regions in both NTWD and CTWD,with variations in three and four amino acid residues,respectively.In total,ten members showed divergence in the WRKY domain,of which seven belonged to group IIc(Table S3).
In addition to the variations in amino acid residues in the WRKY DNA binding domain,some mutations were discovered in the zinc finger motif regions.Four members,including WRKY35 and WRKY114 in group I and WRKY108 and WRKY109 in group III,exhibited variations in amino acid residues in this motif(Table S4).
3.3.Cloning and expression analysis of WRKY genes from upland cotton
By designing gene-specific primers (Table S5),we performed PCR cloning of WRKY genes and amplified the transcripts in given tissues of G.hirsutum acc.TM-1.Finally,we obtained 51 cDNA sequences of WRKY genes with complete ORFs(GenBank accession numbers: KF031069–KF031119),and other genes with partial cDNA sequences.Of the 51 WRKY genes containing complete ORFs,six belonged to group I,37 belonged to group II (4 in group IIa,7 in group IIb,12 in group IIc,8 in group IId,and 6 in group IIe),and eight belonged to group III.
It is well known that the allotetraploid cotton species were formed by an allopolyploidization event occurring approximately 1–2 million years ago,involving a D-genome species as the pollen parent and an A-genome species as the maternal parent[44].We designed gene-specific(not homeolog-specific for the A-and D-subgenomes qRT-PCR primers,Table S5) to evaluate the expression levels of WRKY genes in tetraploid cotton.In total,we detected the expression patterns of 47 WRKY genes in different tissues and organs of G.hirsutum acc.TM-1 by qRT-PCR.These tissues and organs included roots,stems,leaves,petals,anthers,ovules,and fibers at different developmental stages,including 0,10,and 21 DPA.Among the 47 genes examined,12 genes belonged to group I,29 to group II(3 in group IIa,6 in group IIb,8 in group IIc,6 in group IId,and 6 in group IIe),and 6 to group III.These results indicate that WRKY genes from different groups show diverse expression patterns in different tissues and organs.In group I,the expression of the twelve surveyed WRKY genes could be divided into two types,with nine occurring predominantly in reproductive organs and three in vegetative organs(Fig.3).Of these,the transcripts of five genes,WRKY18,WRKY36,WRKY39,WRKY50,and WRKY120,were expressed preferentially in fiber tissues.WRKY22 showed higher expression levels in roots and WRKY59 in leaves.These results suggest that genes belonging to the same domain type have diverse functions.In group II(with five subgroups),the three surveyed WRKY genes in group IIa showed preferential expression in vegetative organs,with the highest level in leaves(Fig.4-A).In group IIb,the expression of six surveyed WRKY genes showed functional diversity,with preferential expression of WRKY54 and WRKY91 in roots,WRKY55 and WRKY58 in fibers,and WRKY45 and WRKY80 in both vegetative and reproductive organs(Fig.4-B).The expression of the eight genes in group IIc also showed functional diversity,with the predominant expression of three genes in roots,three in reproductive organs,and two in both vegetative and reproductive organs(Fig.4-C).These results may be associated with the high structural variation in the WRKY domain regions in group IIc.In group IId,the six surveyed WRKY genes were expressed in all tissues tested,with predominant expression in both vegetative and reproductive organs (Fig.4-D).In group IIe,all six surveyed WRKY genes showed preferential expression in roots,indicating the functional specificity of WRKY genes in this subgroup (Fig.4-E).In group III,the six surveyed WRKY genes all showed preferential expression in vegetative organs,with the preferential expression of three genes in stems,two in roots,and one in leaves(Fig.5).

Fig.6-Expression patterns of WRKY genes under drought stress.The Y-axis indicates the relative expression levels;0,2,4,6,8,10,12,and 24(X-axis)indicate hours of PEG-6000(20%)treatment.The error bars were calculated based on three biological replicates using standard deviation.*significant difference at P <0.05,**means very significant difference(P <0.01).A-G indicate WRKY genes from group I,group IIa,group IIb,group IIc,group IId,group IIe,and group III,respectively.

Fig.7-Expression patterns of WRKY genes under salt stress.The Y-axis indicates relative expression levels and the X-axis the hours of treatment with NaCl (200 mmol L-1).The error bars were calculated based on three biological replicates using standard deviation.* and **: significant difference at P < 0.05 and P < 0.01,respectively.A-E: WRKY genes from group I,group IIa,group IIb,group IIe,and group III,respectively.

Fig.8-Expression patterns of WRKY genes after pathogen inoculation.The Y-axis indicates relative expression levels;0,24,48,96,and 144 h(X-axis)indicate the hours of treatment with a V.dahliae strain VD8 conidial suspension of 107 spores mL-1.The error bars were calculated based on three biological replicates using standard deviation.* and**:significant difference P <0.05 and P <0.01,respectively.A-G:WRKY genes from group I,group IIa,group IIb,group IIc,group IId,group IIe,and group III,respectively.
We further examined the expression of genes that were expressed predominantly in a given organ.Eight genes,including WRKY12,WRKY30,WRKY43,WRKY54,WRKY60,WRKY82,WRKY91,and WRKY110,were expressed predominantly in roots,whereas one gene,WRKY46,was expressed only in stems,two genes,WRKY44 and WRKY59,were expressed only in anthers,and WRKY58 and WRKY55 were expressed only in fibers 10 and 21 DPA,respectively.
3.4.Expression analysis of WRKY genes under different stress conditions
To determine which WRKY genes were induced by different stressors,we performed real-time RT-PCR under three different stress conditions: salt and drought stress (using G.hirsutum cv.Jinmian 19) and V.dahliae (VD) inoculation(using G.barbadense cv.Hai 7124).Sixteen WRKY genes were significantly induced under drought treatment,with six in group I,seven in group II (two in group IIa,one in group IIb,one in group IIc,one in group IId,and two in group IIe),and three in group III (Fig.6).WRKY120 exhibited higher levels of expression at 4 h after drought induction,while the transcripts of other 15 WRKY genes were significantly increased under drought stress,with a peak at 8 h or 10 h of treatment.

Table 2-Expression profile of WRKY genes under different stresses in cotton.
Under salt treatment,12 WRKY genes were significantly induced,including five in group I,four in group II (two in group IIa,one in group IIb,and one in group IIe),and three in group III (Fig.7).The transcripts of five genes in group I and WRKY93 in group IIe were significantly increased under salt treatment,with a peak at 8 or 10 h of treatment.However,the transcripts of other six genes,including three in group II and three in group III,accumulated more quickly and to a higher level at 2 h or 4 h of treatment.
After VD inoculation,fourteen genes were significantly induced,including two in group I,nine in group II (two in group IIa,one in group IIb,three in group IIc,two in group IId,and one in group IIe),and three in group III(Fig.8).There was a rapid and transient induction of the WRKY39 and WRKY93 transcripts,with a peak at 24 h post-inoculation.The transcripts of WRKY41 were significantly upregulated at 24,48,and 144 h post-inoculation,with the highest peak at 48 h of treatment.The transcripts of the other 11 WRKY genes increased significantly in response to inoculation,with a peak at 144 h post-inoculation.
Integrating the expression results of WRKY genes in response to three different stress conditions (Table 2),the expression of eleven WRKY genes was simultaneously induced by both salt and drought.Among them,WRKY46 transcripts showed the highest induced expression after stress treatment.Compared to the untreated control,WRKY46 transcripts accumulated more quickly 4 h to 10 h after drought treatment,with the highest expression(40-fold)at 8 h after treatment.WRKY46 transcripts also accumulated quickly,at 2 h to 12 h after salt treatment,with the highest expression (70-fold) at 4 h,in comparison with the untreated control.This result suggests that WRKY46 plays important roles in the regulation of cotton abiotic stresses such as drought and salt stress.Furthermore,the expression of six WRKY genes,including WRKY59 in group I,WRKY24 and WRKY40 in group IIa,WRKY80 in group IIb,WRKY93 in group IIe,and WRKY64 in group III,was simultaneously induced by the three stressors (drought,salt,and V.dahliae inoculation),suggesting that these WRKY genes function in the regulation of plant stress responses.
4.Discussion
4.1.WRKY gene family evolution and expansion in Gossypium
Cotton,in the genus Gossypium,is the world's most important fiber crop plant.WRKY proteins are members of a transcription factor family in higher plants that play diverse roles in plant responses to various physiological processes.In this study,based on sequence comparison and phylogenetic and structural analysis,we classified WRKY transcription factors in Gossypium into three groups (groups I,II,III),and group II genes were further classified into five subgroups(group IIa–e).Phylogenetic analysis showed that genes in group IIa and group IIb are closely related and that group IId genes are clustered with group IIe.These results support the classifications of the three subgroups,group IIa + group IIb,group IIc,and group IId + group IIe in group II [6,45].Genes in group IIc shared more variations (80%) than genes in other WRKY groups,suggesting that WRKY genes in group IIc are more active and variable than genes in other group II subgroups.
Amplification of the WRKY gene family is also related to species evolution.Zhang et al.[6] reported that numerous duplications and diversifications of WRKY genes,particularly group III genes,have occurred since the divergence of monocots and dicots.In comparison to the 12 members of group III in G.raimondii,there are 14 and 36 group III genes in Arabidopsis and rice,respectively.These are important differences in the number of WRKY genes in dicots versus monocots.
Genome-wide analysis of the WRKY gene family showed that genome duplication contributed to the accumulation of WRKY members.The previous studies reported that there were 72 WRKY family members in Arabidopsis[4],104 members in P.trichocarpa [27],and 57 members in Vitis vinifera (http://www.phytozome.net/).In this study,we identified 120 members of the WRKY gene family in G.raimondii.The genome size of Arabidopsis is 125 Mb [46],whereas the genome sizes of P.trichocarpa,V.vinifera,and G.raimondii are 480.0,487.0,and 737.8 Mb,respectively[32,46–48].WRKY gene family expansion may arise from whole-genome duplication events,rather than from genome size,given that the grapevine genome has not undergone recent genome duplication[48].The Populus genome has undergone salicoid duplication(p event)[47,49],duplication events (β,α) have occurred in Arabidopsis and Gossypium,and Gossypium has undergone one more duplication event than Arabidopsis[49–51].
4.2.WRKY gene family is a superfamily with functional diversity
The WRKY family,one of the most important transcription factor families,regulates plant responses to various physiological processes,especially biotic and abiotic stresses[45,52].Under salt stress,26 WRKY genes were induced in Arabidopsis,based on comprehensive microarray analysis of the root transcriptome [53].Of the 64 GmWRKY genes in soybean(Glycine max Merr.),25 WRKY genes show differential expression in response to at least one abiotic treatment [15].In rice,at least 54 WRKY genes respond to abiotic stress [54].In addition,the transcripts of 49 WRKY genes in Arabidopsis are expressed in response to bacterial infection and salicylic acid(SA) treatment [55].In cotton,eight WRKY genes from different cotton species have previously been reported.GaWRKY1 participates in the regulation of sesquiterpene biosynthesis in cotton,and GhWRKY3 may function in plant defense responses [19,56].In the present study,we further identified 12 WRKY genes induced by salt stress,16 induced by drought stress,and 14 induced in response to V.dahliae VD8 infection.As shown in Table 2,11 WRKY genes were simultaneously induced by both drought and salt treatment,and six WRKY genes were simultaneously induced by drought,salt,and pathogen treatments.These results indicate that WRKY genes are important regulators in cotton stress responses.Notably,GhWRKY59 and GhWRKY80 exhibited sustained responses to V.dahliae inoculation from 48 h to 144 h.They are two of the six WRKY genes simultaneously induced by the three stressors (drought,salt,and V.dahliae inoculation).This finding indicates that GhWRKY59 and GhWRKY80 have multi-functional roles in stress tolerance,and may potentially be applied in breeding for new cotton cultivars with increased stress resistance.
Homologous genes from different plant species may play diverse roles.In Arabidopsis,WRKY genes(AtWRKY2,AtWRKY17,and AtWRKY33) are induced under NaCl treatment [53,57,58],whereas AtWRKY63 may function in drought tolerance[59]and AtWRKY4 and AtWRKY60 function in plant responses to pathogens [7,60].Genes homologous to all of these Arabidopsis WRKY genes except AtWRKY63 were identified in cotton.According to qRT-PCR analysis,WRKY22 and WRKY41,which are homologous to AtWRKY33 and AtWRKY17,respectively,were downregulated in response to NaCl treatment but significantly upregulated under drought treatment and post-inoculation.These results suggest that these homologous genes have different functions in different plant species.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.cj.2014.03.001.
This program was financially supported in part by the National Science Foundation of China(31171590),the Specialized Research Fund for the Doctoral Program of Higher Education of China (20090097110010),the Natural Science Foundation of Jiangsu Province,China (BK2010065),and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
[1] S.Ishiguro,K.Nakamura,Characterization of a cDNA encoding a novel DNA-binding protein,SPF1,that recognizes SP8 sequences in the 5′upstream regions of genes coding for sporamin and beta-amylase from sweet potato,Mol.Gen.Genet.244 (1994) 563–571.
[2] P.J.Rushton,H.Macdonald,A.K.Huttly,C.M.Lazarus,R.Hooley,Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes,Plant Mol.Biol.29 (1995) 691–702.
[3] S.de Pater,V.Greco,K.Pham,J.Memelink,J.Kijne,Characterization of a zinc-dependent transcriptional activator from Arabidopsis,Nucleic Acids Res.24(1996)4624–4631.
[4] T.Eulgem,P.J.Rushton,S.Robatzek,I.E.Somssich,The WRKY superfamily of plant transcription factors,Trends Plant Sci.5(2000) 199–206.
[5] K.L.Wu,Z.J.Guo,H.H.Wang,J.Li,The WRKY family of transcription factors in rice and Arabidopsis and their origins,DNA Res.12(2005) 9–26.
[6] Y.Zhang,L.Wang,The WRKY transcription factor superfamily:its origin in eukaryotes and expansion in plants,BMC Evol.Biol.5(2005) 1.
[7] X.Xu,C.Chen,B.Fan,Z.Chen,Physical and functional interactions between pathogen-induced Arabidopsis WRKY18,WRKY40,and WRKY60 transcription factors,Plant Cell 18(2006) 1310–1326.
[8] T.Eulgem,I.E.Somssich,Networks of WRKY transcription factors in defense signaling,Curr.Opin.Plant Biol.10(2007)366–371.
[9] S.Encinas-Villarejo,A.M.Maldonado,F.Amil-Ruiz,B.de los Santos,F.Romero,F.Pliego-Alfaro,J.Muñoz-Blanco,J.L.Caballero,Evidence for a positive regulatory role of strawberry(Fragaria × ananassa)Fa WRKY1 and Arabidopsis At WRKY75 proteins in resistance,J.Exp.Bot.60(2009)3043–3065.
[10] S.Jing,X.Zhou,Y.Song,D.Yu,Heterologous expression of OsWRKY23 gene enhances pathogen defense and dark-induced leaf senescence in Arabidopsis,Plant Growth Regul.58(2009) 181–190.
[11] S.P.Pandey,I.E.Somssich,The role of WRKY transcription factors in plant immunity,Plant Physiol.150(2009)1648–1655.
[12] Z.Tao,H.Liu,D.Qiu,Y.Zhou,X.Li,C.Xu,S.Wang,A pair of allelic WRKY genes play opposite roles in rice-bacteria interactions,Plant Physiol.151 (2009) 936–948.
[13] P.Abbruscato,T.Nepusz,L.Mizzi,M.Del Corvo,P.Morandini,I.Fumasoni,C.Michel,A.Paccanaro,E.Guiderdoni,U.Schaffrath,J.B.Morel,P.Piffanelli,O.Faivre-Rampant,OsWRKY22,a monocot WRKY gene,plays a role in the resistance response to blast,Mol.Plant Pathol.13(2012)828–841.
[14] N.Ishihama,H.Yoshioka,Post-translational regulation of WRKY transcription factors in plant immunity,Curr.Opin.Plant Biol.15 (2012) 431–437.
[15] Q.Y.Zhou,A.G.Tian,H.F.Zou,Z.M.Xie,G.Lei,J.Huang,C.M.Wang,H.W.Wang,J.S.Zhang,S.Y.Chen,Soybean WRKY-type transcription factor genes,GmWRKY13,GmWRKY21,and GmWRKY54,confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants,Plant Biotechnol.J.6 (2008)486–503.
[16] C.Mare,E.Mazzucotelli,C.Crosatti,E.Francia,A.M.Stanca,L.Cattivelli,Hv-WRKY38:a new transcription factor involved in cold-and drought-response in barley,Plant Mol.Biol.55(2004) 399–416.
[17] S.Li,Q.Fu,L.Chen,W.Huang,D.Yu,Arabidopsis thaliana WRKY25,WRKY26,and WRKY33 coordinate induction of plant thermotolerance,Planta 233 (2011) 1237–1252.
[18] C.S.Johnson,B.Kolevski,D.R.Smyth,TRANSPARENT TESTA GLABRA2,a trichome and seed coat development gene of Arabidopsis,encodes a WRKY transcription factor,Plant Cell 14 (2002) 1359–1375.
[19] Y.H.Xu,J.W.Wang,S.Wang,J.Y.Wang,X.Y.Chen,Characterization of GaWRKY1,a cotton transcription factor that regulates the sesquiterpene synthase gene(+)-delta-cadinene synthase-A,Plant Physiol.135(2004)507–515.
[20] C.Sun,S.Palmqvist,H.Olsson,M.Boren,S.Ahlandsberg,C.Jansson,A novel WRKY transcription factor,SUSIBA2,participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter,Plant Cell 15(2003) 2076–2092.
[21] M.Lagace,D.P.Matton,Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum chacoense,Planta 219 (2004) 185–189.
[22] M.Luo,E.S.Dennis,F.Berger,W.J.Peacock,A.Chaudhury,MINISEED3(MINI3),a WRKY family gene,and HAIKU2(IKU2),a leucine-rich repeat (LRR)KINASE gene,are regulators of seed size in Arabidopsis,Proc.Natl.Acad.Sci.U.S.A.102 (2005)17531–17536.
[23] S.Robatzek,I.E.Somssich,A new member of the Arabidopsis WRKY transcription factor family,AtWRKY6,is associated with both senescence-and defence-related processes,Plant J.28 (2001) 123–133.
[24] S.Robatzek,I.E.Somssich,Targets of AtWRKY6 regulation during plant senescence and pathogen defense,Genes Dev.16 (2002) 1139–1149.
[25] Y.Miao,A.Smykowski,U.Zentgraf,A novel upstream regulator of WRKY53 transcription during leaf senescence in Arabidopsis thaliana,Plant Biol.(Stuttg) (Suppl.-1) (2008)110–120.
[26] J.Ling,W.Jiang,Y.Zhang,H.Yu,Z.Mao,X.Gu,S.Huang,B.Xie,Genome-wide analysis of WRKY gene family in Cucumis sativus,BMC Genomics 12 (2011) 471.
[27] H.He,Q.Dong,Y.Shao,H.Jiang,S.Zhu,B.Cheng,Y.Xiang,Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa,Plant Cell Rep.31(2012)1199–1217.
[28] S.Huang,Y.Gao,J.Liu,X.Peng,X.Niu,Z.Fei,S.Cao,Y.Liu,Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum,Mol.Genet.Genomics 287 (2012)495–513.
[29] H.J.Kim,B.A.Triplett,Cotton fiber growth in planta and in vitro.Models for plant cell elongation and cell wall biogenesis,Plant Physiol.127 (2001) 1361–1366.
[30] F.Yu,Y.Huaxia,W.Lu,C.Wu,X.Cao,X.Guo,GhWRKY15,a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.),is involved in disease resistance and plant development,BMC Plant Biol.12(2012)144.
[31] N.Zhang,P.Zhao,F.Shen,Cloning and expression analysis of 3 WRKY genes from Upland cotton,Mol.Plant Breed.10(2012) 169–173.
[32] A.H.Paterson,J.F.Wendel,H.Gundlach,H.Guo,J.Jenkins,D.Jin,D.Llewellyn,K.C.Showmaker,S.Shu,J.Udall,M.J.Yoo,R.Byers,W.Chen,A.Doron-Faigenboim,M.V.Duke,L.Gong,J.Grimwood,C.Grover,K.Grupp,G.Hu,T.H.Lee,J.Li,L.Lin,T.Liu,B.S.Marler,J.T.Page,A.W.Roberts,E.Romanel,W.S.Sanders,E.Szadkowski,X.Tan,H.Tang,C.Xu,J.Wang,Z.Wang,D.Zhang,L.Zhang,H.Ashrafi,F.Bedon,J.E.Bowers,C.L.Brubaker,P.W.Chee,S.Das,A.R.Gingle,C.H.Haigler,D.Harker,L.V.Hoffmann,R.Hovav,D.C.Jones,C.Lemke,S.Mansoor,M.ur Rahman,L.N.Rainville,A.Rambani,U.K.Reddy,J.K.Rong,Y.Saranga,B.E.Scheffler,J.A.Scheffler,D.M.Stelly,B.A.Triplett,A.Van Deynze,M.F.Vaslin,V.N.Waghmare,S.A.Walford,R.J.Wright,E.A.Zaki,T.Zhang,E.S.Dennis,K.F.Mayer,D.G.Peterson,D.S.Rokhsar,X.Wang,J.Schmutz,Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres,Nature 492(2012) 423–427.
[33] K.Wang,Z.Wang,F.Li,W.Ye,J.Wang,G.Song,Z.Yue,L.Cong,H.Shang,S.Zhu,C.Zou,Q.Li,Y.Yuan,C.Lu,H.Wei,C.Gou,Z.Zheng,Y.Yin,X.Zhang,K.Liu,B.Wang,C.Song,N.Shi,R.J.Kohel,R.G.Percy,J.Z.Yu,Y.X.Zhu,J.Wang,S.Yu,The draft genome of a diploid cotton Gossypium raimondii,Nat.Genet.44(2012) 1098–1103.
[34] Y.X.Zhu,F.G.Li,The Gossypium raimondii genome,a huge leap forward in cotton genomics,J.Integr.Plant Biol.55(2013)570–571.
[35] S.R.Eddy,Accelerated profile HMM searches,PLoS Comput.Biol.7(2011) e1002195.
[36] R.D.Finn,A.Bateman,J.Clements,P.Coggill,R.Y.Eberhardt,S.R.Eddy,A.Heger,K.Hetherington,L.Holm,J.Mistry,E.L.Sonnhammer,J.Tate,M.Punta,Pfam:the protein families database,Nucleic Acids Res.42(2014) D222–D230.
[37] A.Y.Guo,Q.H.Zhu,X.Chen,J.C.Luo,GSDS: a gene structure display server,Hereditas 29 (2007) 1023–1026 (Beijing).
[38] T.L.Bailey,N.Williams,C.Misleh,W.W.Li,MEME:discovering and analyzing DNA and protein sequence motifs,Nucleic Acids Res.34(2006) W369–W373.
[39] J.D.Thompson,T.J.Gibson,F.Plewniak,F.Jeanmougin,D.G.Higgins,The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools,Nucleic Acids Res.25(1997) 4876–4882.
[40] K.Tamura,D.Peterson,N.Peterson,G.Stecher,M.Nei,S.Kumar,MEGA5: molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance,and maximum parsimony methods,Mol.Biol.Evol.28(2011)2731–2739.
[41] J.Jang,T.Zhang,Extraction of total RNA in cotton tissues with CTAB/acidic phenolic method,Cotton Sci.15 (2003)166–167.
[42] K.J.Livak,T.D.Schmittgen,Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT,Methods 25(2001) 402–408.
[43] L.Zhao,Y.Lv,C.Cai,X.Tong,X.Chen,W.Zhang,H.Du,X.Guo,W.Guo,Toward allotetraploid cotton genome assembly:integration of a high-density molecular genetic linkage map with DNA sequence information,BMC Genomics 13(2012)539.
[44] J.Wendel,New World tetraploid cottons contain Old World cytoplasm,Proc.Natl.Acad.Sci.U.S.A.86(1989) 4132–4136.
[45] P.J.Rushton,I.E.Somssich,P.Ringler,Q.J.Shen,WRKY transcription factors,Trends Plant Sci.15(2010) 247–258.
[46] T.A.G.Initiative,Analysis of the genome sequence of the flowering plant Arabidopsis thaliana,Nature 408 (2000)796–815.
[47] G.A.Tuskan,S.Difazio,S.Jansson,J.Bohlmann,I.Grigoriev,U.Hellsten,N.Putnam,S.Ralph,S.Rombauts,A.Salamov,J.Schein,L.Sterck,A.Aerts,R.R.Bhalerao,R.P.Bhalerao,D.Blaudez,W.Boerjan,A.Brun,A.Brunner,V.Busov,M.Campbell,J.Carlson,M.Chalot,J.Chapman,G.L.Chen,D.Cooper,P.M.Coutinho,J.Couturier,S.Covert,Q.Cronk,R.Cunningham,J.Davis,S.Degroeve,A.Déjardin,C.Depamphilis,J.Detter,B.Dirks,I.Dubchak,S.Duplessis,J.Ehlting,B.Ellis,K.Gendler,D.Goodstein,M.Gribskov,J.Grimwood,A.Groover,L.Gunter,B.Hamberger,B.Heinze,Y.Helariutta,B.Henrissat,D.Holligan,R.Holt,W.Huang,N.Islam-Faridi,S.Jones,M.Jones-Rhoades,R.Jorgensen,C.Joshi,J.Kangasjärvi,J.Karlsson,C.Kelleher,R.Kirkpatrick,M.Kirst,A.Kohler,U.Kalluri,F.Larimer,J.Leebens-Mack,J.C.Leplé,P.Locascio,Y.Lou,S.Lucas,F.Martin,B.Montanini,C.Napoli,D.R.Nelson,C.Nelson,K.Nieminen,O.Nilsson,V.Pereda,G.Peter,R.Philippe,G.Pilate,A.Poliakov,J.Razumovskaya,P.Richardson,C.Rinaldi,K.Ritland,P.Rouzé,D.Ryaboy,J.Schmutz,J.Schrader,B.Segerman,H.Shin,A.Siddiqui,F.Sterky,A.Terry,C.J.Tsai,E.Uberbacher,P.Unneberg,J.Vahala,K.Wall,S.Wessler,G.Yang,T.Yin,C.Douglas,M.Marra,G.Sandberg,Y.Van de Peer,D.Rokhsar,The genome of black cottonwood,Populus trichocarpa(Torr.&Gray),Science 313 (2006) 1596–1604.
[48] O.Jaillon,J.M.Aury,B.Noel,A.Policriti,C.Clepet,A.Casagrande,N.Choisne,S.Aubourg,N.Vitulo,C.Jubin,A.Vezzi,F.Legeai,P.Hugueney,C.Dasilva,D.Horner,E.Mica,D.Jublot,J.Poulain,C.Bruyère,A.Billault,B.Segurens,M.Gouyvenoux,E.Ugarte,F.Cattonaro,V.Anthouard,V.Vico,C.Del Fabbro,M.Alaux,G.Di Gaspero,V.Dumas,N.Felice,S.Paillard,I.Juman,M.Moroldo,S.Scalabrin,A.Canaguier,I.Le Clainche,G.Malacrida,E.Durand,G.Pesole,V.Laucou,P.Chatelet,D.Merdinoglu,M.Delledonne,M.Pezzotti,A.Lecharny,C.Scarpelli,F.Artiguenave,M.E.Pè,G.Valle,M.Morgante,M.Caboche,A.F.Adam-Blondon,J.Weissenbach,F.Quétier,P.Wincker,French–Italian Public Consortium for Grapevine Genome Characterization,the grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla,Nature 449 (2007) 463–467.
[49] H.Tang,J.E.Bowers,X.Wang,R.Ming,M.Alam,A.H.Paterson,Synteny and collinearity in plant genomes,Science 320 (2008) 486–488.
[50] J.E.Bowers,B.A.Chapman,J.Rong,A.H.Paterson,Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events,Nature 422 (2003) 433–438.
[51] K.L.Adams,J.F.Wendel,Polyploidy and genome evolution in plants,Curr.Opin.Plant Biol.8 (2005) 135–141.
[52] B.Ulker,I.E.Somssich,WRKY transcription factors:from DNA binding towards biological function,Curr.Opin.Plant Biol.7(2004) 491–498.
[53] Y.Jiang,M.K.Deyholos,Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes,BMC Plant Biol.6 (2006) 25.
[54] R.Ramamoorthy,S.Y.Jiang,N.Kumar,P.N.Venkatesh,S.Ramachandran,A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments,Plant Cell Physiol.49(2008)865–879.
[55] J.Dong,C.Chen,Z.Chen,Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response,Plant Mol.Biol.51(2003) 21–37.
[56] R.Guo,F.Yu,Z.Gao,H.An,X.Cao,X.Guo,GhWRKY3,a novel cotton (Gossypium hirsutum L.) WRKY gene,is involved in diverse stress responses,Mol.Biol.Rep.38(2011) 49–58.
[57] W.Jiang,D.Yu,Arabidopsis WRKY2 transcription factor may be involved in osmotic stress response,Acta Bot.Yunnanica 5(2009) 427–432.
[58] Y.Jiang,M.K.Deyholos,Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses,Plant Mol.Biol.69(2009)91–105.
[59] X.Ren,Z.Chen,Y.Liu,H.Zhang,M.Zhang,Q.Liu,X.Hong,J.K.Zhu,Z.Gong,ABO3,a WRKY transcription factor,mediates plant responses to abscisic acid and drought tolerance in Arabidopsis,Plant J.63 (2010) 417–429.
[60] Z.Lai,K.Vinod,Z.Zheng,B.Fan,Z.Chen,Roles of Arabidopsis WRKY3 and WRKY4 transcription factors in plant responses to pathogens,BMC Plant Biol.8 (2008) 68.
杂志排行

The Crop Journal的其它文章
- Development of highly glyphosate-tolerant tobacco by coexpression of glyphosate acetyltransferase gat and EPSPS G2-aroA genes
- SSR genetic linkage map construction of pea (Pisum sativum L.) based on Chinese native varieties
- Rank correlation among different statistical models in ranking of winter wheat genotypes,
- Effects of exogenous ABA application on post-anthesis dry matter redistribution and grain starch accumulation of winter wheat with different staygreen characteristics
- Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize(Zea mays L.)
- Overexpression of GmDREB1 improves salt tolerance in transgenic wheat and leaf protein response to high salinity