MoV 系复合多金属氧化物催化剂用于丙烷氧化制取丙烯酸(腈)的研究进展
2014-03-10李来平蒋丽娟林小辉
张 新,李来平,刘 燕,蒋丽娟,林小辉,曹 亮,梁 静
(西北有色金属研究院,陕西 西安 710016)
0 前言
丙烯腈和丙烯酸在合成化工材料中十分重要,是关键的有机化工原料[1]。目前丙烯腈和丙烯酸的合成原料主要为丙烯。但丙烯成本较高,随着石油资源的日渐枯竭和天然气资源的大量开发利用,采用价格相对低廉的丙烷选择氧化制备丙烯酸(腈)成本低,工艺路线短,能耗低,不仅能带来巨大的经济效益,而且也更为环境友好,具有十分诱人的应用前景,对低碳烷烃的选择氧化引起了人们愈来愈多的关注。
由于烷烃没有单电子和空轨道的结构特点以及C—H 键较高的键能,使得活化C—H 键比活化C—C 键需要更多的能量,因而不可避免导致C—C 键断裂,引发一系列副反应,从而降低了目标产物的选择性。发展低碳烷烃选择氧化的高效催化剂体系一直是个挑战性的课题[2]。
Mo 组分是丙烷氧化制取丙烯酸(腈)的必需元素,迄今所有报道的相关复合金属氧化物催化剂均含Mo 元素,而且大多数还含有V。MoV 系列复合多金属氧化物催化剂是该反应最重要的一类催化剂。本文就这类催化剂的作用机理和应用等最新研究进展进行讨论。
1 催化剂作用机理研究进展
Botella 等[3]提出在复合金属氧化物催化剂上丙烷氧化存在2 条主要反应途径(见图1):一是烯丙基的氢受亲核晶格氧攻击生成烯丙醇;二是丙烯与催化剂表面BrÊnsted 酸中心反应生成异丙醇盐。烯丙基醇很快转换为丙烯醛,并进一步转换为丙烯酸。异丙醇盐很快转换为丙酮,丙酮经氧化分解又转换为醋酸和Cl 化物,之后与氨作用则生成乙腈、甲腈和氢氰酸。
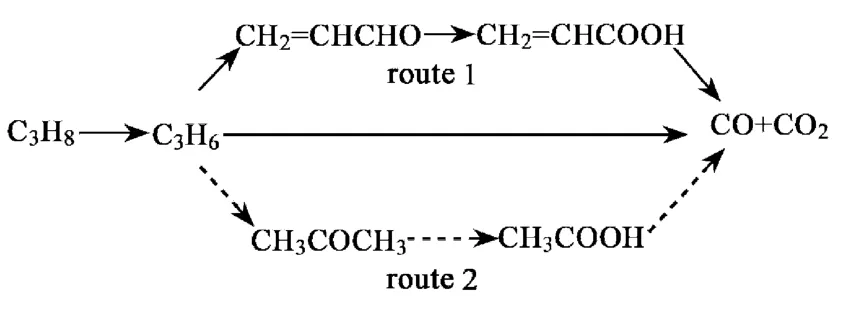
图1 丙烷氧化2 条主要反应途径
Mo 在复合催化剂中的作用主要是用于烯丙基型分子中插入氧,从而使丙烯氧化为丙烯酸:a)烯丙基型分子与氧化物催化剂作用,烃被氧化,氧化物被还原;b)氧化物催化剂重新被气相氧氧化,催化剂复原为起始状态(图2)。
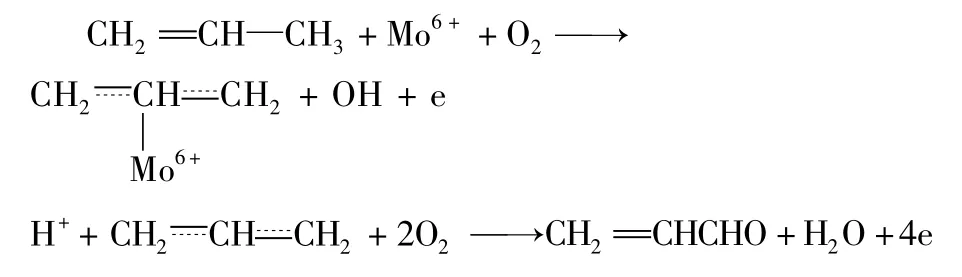
图2 Mo 在复合催化剂中的作用机理
在丙烯氨氧化为丙烯腈的过程中,Mo 是催化剂中重要的活性组分,它具有比较温和的脱氢活性,是丙烯脱氢生成中间体烯丙基的活性中心;由于丙烷氨氧化过程中丙烯腈是通过中间体丙烯生成的,Mo的加入可能会促使中间体更多地向生成丙烯腈的方向进行,从而增大丙烯腈的选择性。
V 是许多烷烃氧化催化剂的关键组分。复合金属氧化物催化剂中正交晶相的形成需要V,且其含量对催化剂性能有着重要影响。Guliants 等[4-5]用LEIS 技术考察了丙烷消耗速度以及丙烯和丙烯酸的生成速度,发现与最外层表面的V 浓度相关,而与Mo 或者Te 的浓度无关,即最外层表面的VOx不但与丙烷的活化、氧化脱氢到丙烯有关,而且与丙烯和丙烯醛中间体进一步氧化有关,其V5+是丙烷反应的活性位[6]。
因此,Mo 和V 是有效催化剂的基本元素组成,由两者参与构成的酸性位可以实现丙烷分子的活化。
在Mo-V 基础上加入Te 和Nb 组分,可以获得丙烷氨氧化至丙烯腈和丙烷氧化至丙烯酸的高效催化剂。Te4+位可以脱除丙烯的α-H,促进中间产物丙烯到丙烯酸的转化并抑制丙烯酸的分解,从而提高了丙烯酸的选择性[7]。Te 的加入也可能使原来的强酸位(其上发生深度氧化)转变为适度酸性的较弱酸位(其上发生选择性氧化)[8]。Nb5+位影响催化剂的氧化还原性,对选择氧化和催化剂再氧化有着重要作用。每个Nb 离子被八面体中的Mo和V 离子分开,隔离了催化剂的活性位[6,9],同时Nb 还有稳定Te 元素的作用,提高表面Te 的浓度而且可以稳定催化剂表面的氧化态和结构。加入Nb组分会改变催化剂的形貌,Mo2V2Te2O 催化剂粒子为长柱状(30~50 μm),而Mo2V2Te2Nb2O 催化剂粒子为短圆柱状(300~500 nm)[10]。吡啶吸附的红外光谱研究表明,加入Nb 组分后B 酸与L 酸位的酸强度和数量均下降,因而抑制了由丙烯到丙酮的平行副反应,并使生成的丙烯酸较快脱附,避免了进一步深度氧化[11]。
另外,在Mo-V 体系中加入Co 也能有效促进催化剂的氧化丙烷至丙烯的选择性。韩智三等[12]研究了Mo-V-Co-O 催化剂对丙烷的氧化脱氢性能,结果表明:添加Co 的催化剂表现出较好的催化性能,在Mo2V1Ox催化剂中添加Co 后,催化剂的可还原性提高,因而催化剂氧化脱氢、迁移和插入晶格氧的能力提高,有利于丙烷活化,提高了丙烷转化率。而且,NH3-TPD 结果表明,在Mo2V1Ox催化剂中添加Co 后,催化剂的酸性降低,碱性增加,有利于催化剂表面丙烯的脱附,从而减少产物丙烯进一步深度氧化,提高了丙烯的选择性[13-14]。
由这几种金属元素氧化物组分即构成了复合多金属氧化物催化剂体系。其中Mo-V-O 催化剂广泛应用于低碳烷烃的催化氧化反应中,Mo-V-Nb-O 具有低温高效催化乙烷氧化脱氢性能;近来报道的Mo-V-Te-Nb-O 催化剂是目前丙烷选择氧化制丙烯酸和氨氧化制丙烯腈最好的催化剂之一[15-17]。
Botella 等研究了Mo-V-Te-Nb-O 系列催化剂的催化反应,并得出了结论。他们认为[18]在催化剂Mo-V-Te-Nb-O 上丙烷选择氧化为丙烯酸存在3 种反应部位:(1)V 位上活化丙烷,如Mo-O-V-O-Mo/Nb;(2)Mo/Te 位上氧化丙烯,如Te-O-Mo-O-Te/V;(3)Mo/Nb 位上氧化丙烯醛至丙烯酸,如Nb-O-Mo-O-Nb/V。这与大部分其他研究者的结论一致,包括Concepción 等[19]认为烷烃氧化要有活化位(V5+)、脱氢位(Te4+,因为有孤对电子)和氧插入位(Mo6+)。
2 丙烷氧化制取丙烯腈催化剂研究进展
丙烯腈生产的关键技术是催化剂,早些时候工程师们采用MoBiCePON 多金属氧化物为氨氧化催化剂,钼等多金属氧化物催化剂中,氧化钼和氧化铋为主催化剂,五氧化二磷为助催化剂,少量氧化铈可抑制丙烯醛的生成,提高催化剂的选择性。
进入21 世纪后,随着丙烯腈需求的增大,世界各国的大型催化剂生产厂家斥巨资研发新型氨氧化催化剂,以提高催化剂的活性、丙烷或异丁烷的转化率和丙烯腈的产率。
目前正在开发中的丙烷氨氧化制丙烯腈催化剂大致可分为3 类。第1 类是锑酸钒催化剂,其通式为VSbmPnAcDbCcOx;第2 类是钒铝氧氮化物(VAl-ON),其通式为VAlxOyNzHn;第3 类是钼酸钒催化剂,即本文中所要介绍的Mo 系催化剂,其通式为Mo1.0VANbbXcZdOn,式中X 表示Te 和Sb 中的一种或两种元素,Z 表示Yb、Dy、Er、Ce、Nd、Sm、La、Pr、Eu、Gd、Tb、Ho、Tm、Lu、Sc、W、Cr、Ta、Ti、Zr、Ha、Mn、Re、Fe、Ru、Co、Rh、Ni、Pd、Pt、Cu、Ag、Zn、B、Al、Ga、In、Ge,Sn、Pb、P、Bi 和碱土金属元素的一种或几种,a=011~110,b=0101~110,c=0101~110,d=0~011[20]。日本三菱化学和旭化成两公司开发的催化剂基本属此类型。
旭化成公司专利中透露的Mo1.0V0.33Nb0.11Te0.22On催化剂的制备方法是将39.0 g 的(NH4)6Mo7O24·4H2O、8.53 g 的NH4VO3和11.16 g H6TeO6溶解于160 g的60 ℃水中,搅拌后冷却至30 ℃得到溶液A。4.25 g Nb2O5·nH2O(Nb2O5质量分数76.0%)和8.27 g 草酸(H2C2O4·2H2O)溶解于50 g 的60 ℃水中,搅拌后冷却至30 ℃得到溶液B。将溶液B 倒入溶液A,并搅拌30 min,得到粗浆混合物。经喷雾干燥后将其置于用聚四氟乙烯涂层的铁板上加热到140 ℃,得到干燥的颗粒状催化剂前体。将该前体装载在内径为20 mm 的石英管中,在氮气氛下600 ℃焙烧2 h 即可[21]。催化剂结构由XRD 等进行表征。
东亚合成株式会社在专利中介绍了一种MoViTejAkOy催化剂的制备方法[22]:将3.64 g 二氧化碲溶于60 mL 水,在80 ℃下加入2.8 g 单水合肼并保持12 h,过滤后得到黑色金属碲晶体粉末并用水稀释至80 mL 分散体。将2.56 g 偏钒酸铵、15.45 g钼酸铵和50 mL 水在沸点下溶解,将一半的含碲水分散体加入溶液热处理1 h 并用冰水冷却至30 ℃。将4.41 g 草酸和1.74 g 铌酸分别溶解在70 mL 水中并添加至上述反应液体,将混合液剧烈搅拌10 min然后加入2.5 g 硝酸铵,混合物加热浓缩并在120 ℃下蒸干,所得物质在空气中300 ℃煅烧5 h,在氮气中600 ℃煅烧2 h,得到催化剂。该催化剂不仅用于制取丙烯腈,也可用于丙烷直接氧化制取丙烯酸。
3 丙烷直接氧化制取丙烯酸研究进展
由丙烷一步氧化制丙烯酸已有一些基础研究报道,采用的催化剂主要包括钒磷氧(V-P-O)、杂多酸及其盐(HPCs)以及复合金属氧化物(MMo)等3 类体系。将工业化生产用的丁烷氧化制马来酸酐的V-P-O 催化剂体系用于丙烷选择氧化制丙烯酸时效果并不理想,单程收率往往不超过19%;杂多酸及其盐虽然具有结构可控的优点,但由于其制备过程中不经过焙烧,催化剂结构不够稳定,在高于400 ℃的操作温度下容易因结构坍塌而失活,而且其对丙烷一步氧化制丙烯酸的效果也不理想,最高的丙烯酸收率仅为13%。从1991 年开始,以MoV基催化剂为代表的混合金属氧化物催化剂体系得到了广泛的研究,在丙烷选择氧化制丙烯酸的反应中也取得了较好的催化效果。日本Mitsubishi-Kasai公司使用Mo-V-Te-Nb-O 催化剂获得了高达52.5%的丙烯酸产率;美国Rohm&Hass 公司报道的丙烯酸收率可达42%。
目前报道的催化剂制备方法包括水热法,旋转蒸发法,物理气相沉积法,固相法以及自动合成法等。由固相法制备的催化剂活性较差。采用水热合成与旋转蒸发法制得的催化剂活性较高,但制备周期长。采用自动合成法制备的催化剂其晶相结构和催化性能有较好的重现性。
祝宝东等[23]采用水溶液法制备Mo-V-Te-Nb-O 催化剂,通过XRD 和BET 对催化剂进行表征,考察连续固定床微型反应研究制备过程中干燥方式、干燥温度及煅烧方式对催化剂结构和性能的影响。结果表明,减压下旋转蒸发与常压下加热蒸发的干燥方式相比,前者利于生成活性组分分布均匀的催化剂前驱体,且催化剂晶相结构中有利于提高活性和选择性的M1相、M2相比例较大,催化剂比表面积比后者增加了31.5%,适宜的干燥温度为50 ℃,N2,He 气氛在催化剂煅烧过程中均起到防止空气氧化的作用,对催化剂的结构和性能影响相同,空气中275 ℃下预煅烧60 min 制备的催化剂晶相结构中Te 含量增加,该催化剂比表面积为2.78 m2/g,在反应温度为360 ℃时表现出良好的催化性能,丙烷的转化率为47.6%,丙烯酸的选择性为50.5%。
Botella 等[24]以水热合成法,用Anderson 型钼碲杂多酸、硫酸氧钒和草酸铌为原料制备的4 组分Mo2V2Te2Nb2O 催化剂,其丙烯酸收率达到36%~38%。水热合成通常包括以下过程:各组分起始原料通常是钼酸铵、硫酸氧钒、二氧化碲(碲酸)和草酸铌(铵)。室温(或加热)下搅拌溶解,调节pH,然后转入水热釜,水热处理一定时间,抽滤,干燥,在550~600 ℃静态惰性气氛(氮气或者氩气)中活化以抑制组分的相分离。Mo 组分在混合液中的浓度对于得到特定的晶相结构至关重要。
Katou 等[25]通过控制水热合成过程中水的加入量,得到结晶型(对应低的Mo 离子浓度)和无定型(对应高的Mo 离子浓度)催化剂,说明制备过程中组分元素的浓度是一个重要的影响因素。
除了非载型催化剂,人们也尝试制备负载型催化剂。李洪波等[26]对比了Al2O3负载AgBiVMoO4型催化剂和非负载型催化剂上丙烷氧化制取丙烯酸的研究。结果表明:负载载体后,催化剂的反应选择性得到了一定改善,气相产物中丙烯的选择性从22.6%提高到27.0%,COx选择性从56.53%降低到45.80%;液相产物的总选择性也从7.78%增加到11.32%。尽管负载型催化剂所含活性组分的质量仅为非负载型催化剂的40%,却表现了较好的反应活性,得到丙烷的转化率接近非负载型催化剂的结果。说明负载了Al2O3载体后,催化剂的反应性能得到了明显改善,更有利于提高液相产品中丙烯酸的选择性。
4 结论与展望
由丙烷直接氧化制取丙烯酸(腈)可以充分利用低碳烷烃资源,简化工艺路线,降低操作成本,而且有利于环境保护,因此在国内外引起人们的广泛兴趣,且已经成为催化剂研究领域的一个热点研究课题。多组分金属氧化物催化剂具有良好的稳定性、有效性和催化选择性。然而回收率不高仍是其最大的难点,无论是制取丙烯酸或者丙烯腈,收率仍只有50%左右,其经济性优势尚不能很好地体现,但是从长远来看,由于石油资源的日益减少和天然气资源的大量开发,丙烷氧化制取丙烯酸(腈)的技术发展前景是十分诱人的。
[1]张文钲.钼基催化剂技术发展现状[J],中国钼业,2011,35(2):1-6.
[2]Kenzo Oshihara,Yasuhiro Nakamura,Mayumi Sakumaa,Wataru Uedab.Hydrothermal synthesis of novel crystalline Mo-V-M-O (M-Al,Ga,Fe)mixed oxide in the presence of triethylammonium chloride and their catalytic performance for selective ethane oxidation[J].Catalysis Today,2001,71:1-2.
[3]Botella P,Solsona B,Martinez-Arias A.Selective oxidation of propane to acrylic acid on MoVNbTe mixed oxides catalysts prepared by hydrothermal synthesis[J].Catal.Lett.,2001,74:149-154.
[4]Guliants V V,Bhandari R.A study of the surface region of the Mo-V-Te-O catalysts for propane oxidation to acrylic acid[J].J .Phys.Chem.B,2005,109:10234-10242.
[5]Guliants V V,Bhandari R,Hughett A R.Probe molecule chemisorption-low energy lon scattering study of surface active sites present in the orthorhombic Mo-V-(Te-Nb)-O catalysts for propane (amm)oxidation[J].J.Phys.Chem.B,2006,110:6129-6140.
[6]Grasselli R K,Burrington J D,Buttrey D J.Multifunctionality of active centers in (amm)oxidation catalysts:from Bi–Mo–Oxto Mo– V– Nb–(Te2Sb)– Ox[J].Top.Catal.,2003,23:5-22.
[7]Lin M.M.Complex metal-oxide catalysts for selective oxidation of propane and derivatives:I.catalysts preparation and application in propane selective oxidation to acrylic acid[J].Appl.Catal.A,2003,250:305-318.
[8]Katou T,Vitry D,Ueda W.Structure dependency of Mo-V-O-based complex oxide catalysts in the oxidations of hydrocarbons[J].Catal.Today,2004,91-92:237-240.
[9]Grasselli R K.Structure dependency of Mo-V-O-based complex oxide catalysts in the oxidations of hydrocarbons[J].Catal.Today,2005,99:23-31.
[10]Vitry D,Morikawa Y,Dubois J L.Mo-V-Te-(Nb)-O mixed metal oxides prepared by hydrothermal synthesis for catalytic selective oxidations of propane and propene to acrylic acid[J].Appl.Catal.,A,2003,251:411-424.
[11]Ueda W,Vitry D,Katou T.Crystalline Mo-V-O based complex oxides as selective oxidation catalysts of propane[J].Catal.Today,2005,99:43-49.
[12]韩智三,伊晓东,林 洪,等.Mo-V-Co-O 催化剂的丙烷氧化脱氢性能研究[J].厦门大学学报,2005,44:67-70.
[13]Sam D S;Soenen V;Volta J C,Oxidation dehydrogenation of propane over V-Mg-O catalysts [J].J.Catal.,1990,123:417-435.
[14]Chen K D,Xie S B,Bell A T.Alkali effects on molybdenum oxide catalyst s for the oxidation dehydrogenation of propane[J].J.Catal.,2000,195 (2):244-252.
[15]Thorsteninson E M,Wison T P,Young F G.Theoxidation dehydrogenation of ethane over catalyst s containing mixed oxides of molybdenum and vanadium [J].J.Catal.,1978,52:116-132.
[16]Burch R,Swarnakar R.Oxidative dehydrogenation of ethane on vanadium-molybdenum oxide and vanadium-ni-bi-um-molybdenum oxide catalysts[J].Appl.Catal.A:Gen.,1991,70:129-135.
[17]Ruth K,Burch R.Mo2V2Nb oxide catalyst s for the partialoxidation of ethane 2II chemical and catalytic-propertiesand structure function relationships[J].J.Catal.,1998,175:27-39.
[18]Botella P,García2González E,Nieto J M L.MoVTeNbO multifunctional catalysts:Correlation between constituent crystalline phases and catalytic performance.Solid State Sci[J].2005,7 :507-519.
[19]Concepción P,Botella P,Nieto J ML.Catalytic and FT-IR study on the reaction pathway for oxidation of propane and propylene on V-or Mo–V-based catalysts[J].Appl.Catal.A,2004,278:45-56.
[20]谢方友.丙烷氨氧化制丙烯腈催化剂设计进展[J].工业催化,2003,11 (8):38-42.
[21]Hinago H,Komada S.Ammoxidation catalyst for use in producing acrylonitrile or methacrylonitrile from propane or isobutene by ammoxidation.美国,US 6063728[P],2000-05-16.
[22]住田勇一,屠新林,新妻裕志,等,金属氧化物催化剂的制备方法.中国,CN 200480010210[P].2006-05-17.
[23]祝宝东,王 鉴,董 群,等.丙烷选择氧化制备丙烯酸Mo-V-Te-Nb-O 催化剂[J].石油化工高等学校学报,2009,22(4):26-29.
[24]Botella P,Nieto J ML,Solsona B.The preparation,characterization,and catalytic behavior of MoVTeNbO catalysts prepared by hydrothermal synthesis[J].J.Catal.,2002,209:445-455.
[25]Katou T,Vitry D,Ueda W.Structure dependency of Mo-V-O-based complex oxide catalysts in the oxidations of hydrocarbons[J].Catal.Today,2004,91-92:237-240.
[26]李洪波,杨 萍,刘 杰,等.负载型催化剂上丙烷选择氧化制丙烯酸的研究[J].精细化工,2003,20(12):728-730.
