超高效液相色谱-同位素稀释质谱法测定配方奶粉中的泛酸
2014-03-09崔亚娟李全霞陈兆天梁敏慧
渠 岩,崔亚娟,李全霞,陈兆天,李 东,梁敏慧
(1.北京市营养源研究所,北京 100050;2.北京工业大学生命科学与生物工程学院,北京 100022)
超高效液相色谱-同位素稀释质谱法测定配方奶粉中的泛酸
渠 岩1,崔亚娟1,李全霞1,陈兆天1,李 东1,梁敏慧2
(1.北京市营养源研究所,北京 100050;2.北京工业大学生命科学与生物工程学院,北京 100022)
建立测定配方奶粉中泛 酸的超高效液相色谱-同位素稀释质谱方法。样品经乙酸铵溶液提取、三氯甲烷除蛋白后,进行超高效液相色谱-串联质谱分析,采用HSS T3液相色谱柱分离,以10 mmol/L乙酸铵溶液(含0.1%甲酸)和乙腈为流动相进行梯度洗脱,多反应监测模式,内标法定量。结果表明:方法定量限为0.040 mg/100 g,线性范围内低、中、高3 个标准添加水平的回收率为96.9%~104.3%,相对标准偏差为3.78%~5.04%。该方法操作过程简单、分析周期短、灵敏度高、重复性好,适用于奶粉中泛酸的测定。
泛酸;超高效液相色谱-同位素稀释质谱方法;配方奶粉
泛酸又称VB5或遍多酸,广泛存在于生物体内,是生理代谢所必需的酸类物质。泛酸在酸、碱、热及光等条件下都不稳定。泛酸是辅酶A的组成部分,参与体内蛋白质、碳水化合物的生理代谢,尤其是对脂肪的合成和代谢起着十分重要的作用[1]。根据《中国居民膳食营养素参考摄入量》[2],泛酸每日适宜摄入量为:婴幼儿1.7~1.8 mg,未成年人2.0~4.0 mg,成年人5.0 mg,孕妇6.0 mg,乳母7.0 mg。
目前,配方奶粉中泛酸的国家标准检测方法是微生物法和高效液相色谱法[3]。其中,微生物法[4-5]是测定泛酸的经典方法,但是该方法复杂费时,对环境条件要求高。高效液相色谱法[6-12]常用来检测强化食品中的泛酸,但其所用的流动相均为磷酸盐缓冲溶液,需要调节溶液pH值,过程复杂且容易受到其他添加剂的影响。除此以外,也有报道用免疫法、气相色谱-质谱联用法、高效毛细管电泳法、超临界流体色谱法等[13-17]方法检测泛酸。超高效液相色谱串联质谱(ultra performance liquid chromatography-tandem mass spectrometry,UPLC-MSMS)法是一种在定性和定量方面都具有优势的方法,具有较高的灵敏度和较强的分离能力,可以在短时间内分析低含量、多组分的样品[18-20]。近年来,国外有报道利用泛酸的稳定同位素结合UPLC-MS-MS法测定食品中的泛酸[21-25],该方法被证实是一种快速、准确、灵敏的方法,然而国内相关研究却鲜有报道。
1 材料与方法
1.1 材料与试剂
泛酸钙(纯度≥98.0%) 美国Supelco公司;泛酸钙-[13C3,15N](纯度≥99.5%) 美国IsoSciences公司;乙酸铵、甲酸(均为色谱纯) 美国Sigma公司;乙腈(色谱纯) 美国Fisher公司;三氯甲烷(分析纯)北京化学试剂公司;实验用水为超纯水。
1.2 仪器与设备
Acquity@UPLC-Xevo TQ 型超高效液相色谱-串联质谱联用仪、Acquity@HSS T3柱 美国Waters公司;高速冷冻离心机 日本Hitachi公司;Vortex-Genie 2型漩涡振荡器 美国Scientific Industries公司;BS224S型分析天平 德国Sartorius公司。
1.3 方法
1.3.1 溶液配制
泛酸标准溶液(100 μg/mL):称取泛酸钙10.87 mg,加水溶解至100 mL(泛酸质量浓度=泛酸钙质量浓度×0.92);泛酸同位素标准溶液(100 μg/mL):称取泛酸钙-[13C3,15N] 1.09 mg,加水溶解至10 mL;10 mmol/L乙酸铵缓冲液:称取乙酸铵0.385 4 g于500 mL容量瓶中,加水溶解并定容。
1.3.2 样品处理
称取奶粉样品约5 g(精确至0.000 1 g)于150 mL三角瓶中,加入100 mL 40~50 ℃温水,振摇溶解后超声萃取10 min。取试样溶液1 mL于50 mL离心管中,加入泛酸同位素标准溶液(由初步实验确定添加量,确保试样中泛酸和同位素泛酸质量相近),用10 mmol/L乙酸铵缓冲液定容至20 mL。涡旋振荡1 min,超声10 min。加入10 mL三氯甲烷,涡旋振荡1 min,10 000 r/min离心10 min。上清液用0.22 μm gHP滤膜过滤,并转移至棕色自动进样瓶中。
1.3.3 色谱条件
色谱柱:HSS T3柱(2.1 mm×100 mm,1.8 μm);流速0.4 mL/min;柱温40 ℃;样品温度15 ℃;进样量10 μL;流动相A:10 mmol/L乙酸铵(含0.1%甲酸)溶液;流动相B:乙腈。梯度洗脱条件见表1。

表1 流动相梯度洗脱条件Table 1 Elution conditions for PA
1.3.4 质谱条件
电喷雾离子源;正离子模式;多反应监测模式;毛细管电压1.5 kV;离子源温度150 ℃;脱溶剂气温度500 ℃;锥孔气流量(氮气)50 L/h;脱溶剂气流量(氮气)1 000 L/h;碰撞气流速(氩气)0.17 mL/min。
泛酸和同位素泛酸的定量和定性离子对、锥孔电压、碰撞能量等参数见表2。
表2 泛酸和同位素泛酸的定性和定量离子对、锥孔电压和碰撞能量Table 2 Qualitative and quantitative ion pairs, cone voltages and collision energy for PA and PA-

表2 泛酸和同位素泛酸的定性和定量离子对、锥孔电压和碰撞能量Table 2 Qualitative and quantitative ion pairs, cone voltages and collision energy for PA and PA-
注:*.定量离子对。
化合物 定性定量离子对(m/z)锥孔电压/V碰撞能量/eV泛酸220.1>90.0*2212 220.1>202.02212泛酸-[13C3,15N]224.1>94.0*2514 224.1>206.02514
2 结果与分析
2.1 样品处理的优化
考察了不同提取剂的提取效果,用水和乙酸铵缓冲液分别提取,泛酸的回收率均在90%~110%之间。但由于不同样品在水中的pH值不同,会影响泛酸在色谱柱上的保留时间和峰形。而利用乙酸铵缓冲液提取可以得到较稳定的保留时间和较好的峰形,因此选择其作为提取剂。
奶粉中含有大量蛋白质,如不除去会使样品混浊、无法进样,更会缩短色谱柱寿命。因此,考察了三氯甲烷、高氯酸和调节pH值的除蛋白效果和对泛酸回收率的影响。结果显示,3种方式都可以使蛋白沉淀,获得澄清的样品溶液。但高氯酸对泛酸的破坏性很大,处理过后的泛酸响应值显著降低。而三氯甲烷和调节pH值这2种方式均对泛酸回收率无显著影响,回收率分别为99.3%和96.3%。考虑到操作简易省时,选择添加三氯甲烷去除蛋白。
2.2 色谱与质谱条件的优化
2.2.1 色谱柱的选择
比较了BEH C18柱(100 mm×2.1 mm,1.7 μm)、BEH Amide柱(100 mm×2.1 mm,1.7 μm)和HSS T3柱(100 mm×2.1 mm,1.8 μm)的分离效果(图1)。因为泛酸属于极性较强的化合物,它在C18柱和Amide柱上的保留较差,峰形也较差。而在对极性化合物有特殊保留能力的HSS T3柱上得到了较好的分离效果,且峰形好,响应值高,因此选择HSS T3柱。

图1 泛酸在3种色谱柱上的离子色谱图Fig.1 Ion chromatograms of PA by using three columns
2.2.2 多反应监测条件的选择
本研究使用UPLC-MS-MS方法,通过多反应监测方式进行采集。选择目标物质的2对母离子>子离子,其中响应值较强的离子对作为定量离子对,另一对作为定性离子对。泛酸和同位素泛酸的多反应监测色谱图、子离子扫描图见图2、3。


图2 泛酸和同位素泛酸的离子色谱图Fig.2 Ion chromatograms of PA and PA-[13C3,15N]

图3 泛酸(A)和同位素泛酸(B)的子离子扫描图Fig.3 Mass spectra of PA (A) and PA-[13C3,15N] (B)
2.3 方法的验证
2.3.1 回收率
取已知泛酸含量的奶粉样品,进行3 个标准添加水平(样品中泛酸含量的50%、100%、150%)的回收率实验,每个添加水平重复测定6次,分别计算加标回收率和相对标准偏差,结果见表3。泛酸3个水平的加标回收率在96.9%~104.3%,相对标准偏差在3.78%~5.04%,能够满足检测实际样品需要。

表3 泛酸加标回收实验结果Table 3 Average recoveries and relative standard deviations of PA
2.3.2 线性范围、检出限和定量限
准确吸取泛酸标准溶液,分别添加泛酸同位素标准溶液,用10 mmol/L乙酸铵缓冲液稀释成质量浓度为1、50、100、200、300、400、500 ng/mL的泛酸标准工作液,以峰面积比(A泛酸/A同位素)和同位素质量浓度的乘积为纵坐标、泛酸质量浓度为横坐标,绘制标准曲线。结果表明,泛酸在1~500 ng/mL范围内线性关系良好,线性回归方程为Y=1.038 56X+0.953 334,相关系数r=0.999 9。根据定量离子信噪比(RSN)结合定量限加标回收实验(回收率92.9%),确定检出限(RSN≥3)为0.4 ng/mL,定量限(RSN≥10)为1.0 ng/mL。当取样量为5 g、最终定容体积为20 mL时,检出限和定量限分别为0.016 mg/100 g和0.040 mg/100 g。
2.3.3 方法精密度
取奶粉样品按照1.3节中的方法,每日平行测定6次(n=6),连续测定3 d(n=3),考察方法的精密度,结果见表4。
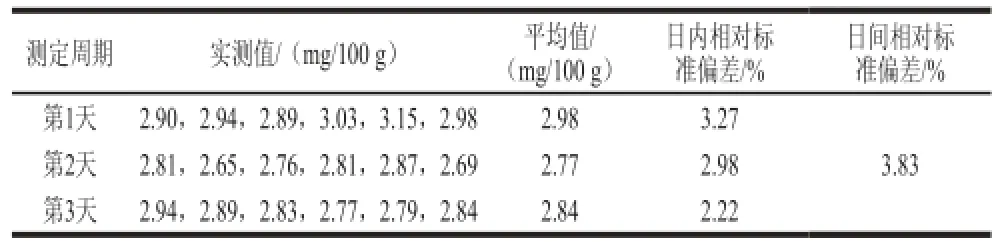
表4 样品中泛酸的测定结果Table 4 Results of PA determination in samples
2.3.4 与微生物方法的比对
微生物法是检测泛酸的经典方法,同时也是GB 5413.17—2010《婴幼儿食品和乳品中泛酸的测定》[3]的第一法。用微生物法和UPLC-MS-MS法同时对10种奶粉样品做比对实验(表5)。对实验结果做t检验:平均差的95%置信区间,假设平均差=0(与平均差≠0),t=-0.519,P=0.309>0.05,表明2种方法测定结果无显著差异,同时也证明了UPLC-MS-MS法测定结果的准确性。
然而由于微生物法操作复杂、培养耗时等因素,导致其检测周期较长(3~4 d),不适于大量样品的快速检测。而UPLC-MS-MS法的样品处理步骤简单、操作省时,在保证分析结果准确的前提下,可以大幅缩短样品的检测周期,有利于提高检测效率。

表5 泛酸含量测定微生物法和UPLC-MS-MS法对比Table 5 Comparison of microbiological method and UPLC-MS-MS mg/100 g
3 结 论
本研究通过对样品前处理方法和UPLC-MS-MS条件优化,建立了一种可以快速检测配方奶粉中泛酸的方法。本方法利用乙酸铵缓冲液提取、采用三氯甲烷除蛋白,得到了较好的提取效果。同时利用泛酸同位素作为内标,通过多反应监测模式进行信号采集,样品回收率较高且稳定。本方法结果准确,与传统的微生物法相比没有显著差异,而且方法操作过程简单、分析周期短、灵敏度高、重复性好,适用于奶粉中泛酸的测定。
[1] 杨延辉, 肖春玲. 泛酸的功能和生物合成[J]. 生命的化学, 2008, 28(4): 448-452.
[2] 中国营养学会. 中国居民膳食营养素参考摄入[M]. 北京: 中国轻工业出版社, 2000: 350-356.
[3] 卫生部. GB 5413.17—2010 婴幼儿食品和乳品中泛酸的测定[S]. 北京: 中国标准出版社, 2010.
[4] ANGYAL G. Methods for microbiological analysis of selected nutrients[J]. Journal of AOAC International, 1996, 8 (1): 41-45.
[5] 张旭, 马妮, 郑洪. 微生物法测定食品中泛酸 的含量[J]. 中国微生态学杂志, 2012, 24(7): 654-655.
[6] 林丽琴, 杨直, 石云峰. HPLC测定泛酸钙的有关物质[J]. 中国现代应用药学, 2012, 29(11): 10135-1038.
[7] 杜彦山, 张志国, 贾云虹, 等. 高效液相色谱法测定奶粉中泛酸[J].食品研究与开发, 2007, 28(6): 121-124.
[8] 刘志楠, 喻东威, 宋晓东, 等. 烟酸和泛酸不同方法检测的对比[J].食品研究与开发, 2011, 32(11): 90-93.
[9] 杨发树, 刘耀敏, 陈照. 反相高效液相色谱法测定预混合饲料中的D-泛酸[J]. 饲料研究, 2012, 35(8): 73-75.
[10] 刘志楠, 喻东威, 赵源, 等. 牛奶中泛酸含量测定[J]. 食品科学, 2012, 33(2): 177-180.
[11] 李少旦, 彭卫芳. 反相高效液相色谱法同时测定维生素B6、烟酰胺和泛酸钙[J]. 理化检验: 化学分册, 2009, 45(7): 800-802.
[12] PAKIN C, BERGAENTZLE M, HUBSCHER V, et al. Fluorimetric determination of pantothenic acid in foods by liquid chromatography with post-column derivatization[J]. Journal of Chromatography A, 2004, 1035(1): 87-95.
[13] HAUGHEY S A, O’KANE A A, BAXTER G A, et al. Determination of pantothenic acid in foods by optical biosensor immunoassay[J]. Journal of AOAC International, 2005, 88(4): 1008-1014.
[14] BANNO K, MATSUOKA M, HORIMOTO S, et al. Simultaneous determination of pantothenic acid and hopantenic acid in biological samples and natural products by gas chromatography-mass fragmentography[J]. Journal of Chromatography B, 1990, 525: 255-264.
[15] RYCHLIK M. Quantification of free and bound pantothenic acid in foods and blood plasma by a stable isotope dilution assay[J]. Journal of Agricultural and Food Chemistry, 2000, 48(4): 1175-1181.
[16] ZHANG Q, QIU J, LIAO N S. Simultaneous determination of calcium pantothenate and calcium-amino propionate and γ-butyrolactone by high performance capillary zone electrophories[J]. Chinese Journal of Analysis Laboratory, 2001, 20(1): 91-92.
[17] 程劼, 谢建春, 苏晓鸥. 饲料中D-泛酸钙的超临界流体色谱测定[J].分析测试学报, 2010, 29(4): 418-420.
[18] LEPORATI A, CATELLANI D, SUMAN M, et al. Application of a liquid chromatography tandem mass spectrometry method to the analysis of water-soluble vitamins in Italian pasta[J]. Analytica Chimica Acta, 2005, 531(1): 87-95.
[19] CHEN Z, CHEN B, YAO S Z. High-performance liquid chromatography/ electrospray ionization-mass spectrometry for simultaneous determination of taurine and 10 water-soluble vitamins in multivitamin tablets[J]. Analytica Chimica Acta, 2006, 569(1): 169-175.
[20] LU B Y, REN Y P, HUANG B F, et al. Simultaneous determination of four water-soluble vitamins in fortified infant foods by ultraperformance liquid chromatography coupled with triple quadrupole mass spectrometry[J]. Journal of Chromatographic Science, 2008, 46(3): 225-232.
[21] RYCHLIK M, FREISLEBEN A. Quantification of pantothenic acid and folates by stable isotope dilution assays[J]. Journal of Food Composition and Analysis, 2002, 15(4): 399-409.
[22] RYCHLIK M. Simultaneous analysis of folic acid and pantothenic acid in foods enriched with vitamins by stable isotope dilution assays[J]. Analytica Chimica Acta, 2003, 495(1/2): 133-141.
[23] RYCHLIK M. Pantothenic acid quantification by a stable isotope dilution assay based on liquid chromatography-tandem mass spectrometry[J]. Analyst, 2003, 128(7): 832-837.
[24] GUTZEIT D, KLAUBERT B, RYCHLIK M, et al. Effects of processing and of storage on the stability of pantothenic acid in sea buckthorn products (Hippophaё rhamnoides L. ssp. rhamnoides) assessed by stable isotope dilution assay[J]. Journal of Agricultural and Food Chemistry, 2007, 55(10): 3978-3984.
[25] RYCHLIK M, ROTH-MAIER D. Pantothenic acid quantification: method comparison of a stable isotope dilution assay and a microbiological assay[J]. International Journal for Vitamin and Nutrition Research, 2005, 75(3): 218-223.
Determination of Pantothenic Acid in Formula Milk Powder Using Ultra Performance Liquid Chromatography-Isotope Dilution Mass Spectrometry
QU Yan1, CUI Ya-juan1, LI Quan-xia1, CHEN Zhao-tian1, LI Dong1, LIANG Min-hui2
(1. Beijing Research Institute for Nutritional Resources, Beijing 100050, China; 2. College of Life Science and Bio-enginee ring, Beijing University of Technology, Beijing 100022, China)
An ultra performance liquid chromatography-isotope dilution mass spectrometry (UPLC-IDMS) method has been developed for the determination for pantothenic acid (PA) in formula milk powder. Samples were extracted with ammonium acetate solution, and precipitated protein by adding chloroform for analyzing by UPLC-MS-MS. The analyte was separated using an HSS T3 column. The mobile phase consisted of 10 mmol/L ammonium acetate with 0.1% formic acid and acetonitrile. The PA was identified by multiple reaction monitoring (MRM) and quantified by internal standard method. The results showed that the limit of quantification for PA was 0.040 mg/100 g. The recoveries at three spiked levels and the relative standard deviations were 96.9%–104.3% and 3.78%–5.04%, respectively. This method proved to be simple, timesaving, sensitive and accurate for the determination of PA in formula milk powder.
pantothenic acid; ultra performance liquid chromatography-isotope dilution mass spectrometry; formula milk powder
TS207.3;O657.63
A
1002-6630(2014)08-0212-05
10.7506/spkx1002-6630-201408042
2013-08-01
北京市科技新星计划项目(2011059);卫生行业科研专项项目(201202012);
北京市科委“双十计划”项目(Z121106002812109);北京市优秀人才培养资助项目(2011D002022000001)
渠岩(1984—),男,工程师,硕士,研究方向为食品分析。E-mail:quyan_0220@163.com
