分散固相萃取-超高效液相色谱-串联质谱检测水产品中14 种喹诺酮类药物
2014-03-08李佩佩张小军梅光明严忠雍郭远明
李佩佩,张小军,梅光明,严忠雍,龙 举,刘 琴,郭远明*
(浙江省海洋水产研究所,浙江省海水增养殖重点实验室,浙江 舟山 316100)
分散固相萃取-超高效液相色谱-串联质谱检测水产品中14 种喹诺酮类药物
李佩佩,张小军,梅光明,严忠雍,龙 举,刘 琴,郭远明*
(浙江省海洋水产研究所,浙江省海水增养殖重点实验室,浙江 舟山 316100)
采用基质固相分散的前处理技术,建立一种超高效液相色谱-串联质谱同时检测水产品中14 种喹诺酮类药物的分析方法。样品用5%甲酸-乙腈溶液提取和盐析剂盐析后加入N-丙基乙二胺和十八烷基键合硅胶吸附剂(C18)净化剂进行基质固相分散净化,氮吹复溶后经Waters ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm)分离,以0.2%甲酸溶液和甲醇为流动相梯度洗脱,采用电喷雾离子源正离子多反应监测模式测定,外标法定量。14 种药物在0.50~20.0 μg/L质量浓度范围内呈良好线性,线性相关系数不小于0.995,方法检出限为0.4~1.0 μg/kg,定量限为1.0~3.0 μg/kg。14 种药物在3 个加标水平下的平均回收率为82%~90%,相对标准偏差(n=5)均小于11.0%。本方法简便快速、灵敏度高、实用性强,可作为水产品中14 种喹诺酮类药物残留的快速确证和定量分析。
基质固相分散;超高效液相色谱-串联质谱;水产品;喹诺酮类药物
喹诺酮(quinolone antibiotics,QNs)是一类高效且价格低廉的广谱抗菌药,广泛应用于畜牧和水产养殖中[1]。随着该类药物的非科学和过量使用,其在动物体内残留后通过食物链进入人体,危害人体健康[2];同时未被吸收的部分随排泄物进入环境,导致形成“假持久性”现象[2],成为新型环境污染物,对人类和环境亦造成严重威胁。因此动物体内的QNs的残留问题备受关注。美国禁止在食品动物养殖中使用QNs[3];国际食品法典委员会、欧盟、日本、我国均制订出相应的最高残留限量:根据不同动物品种、组织和药物种类,QNs的最高残留限量在10~6 000 μg/kg[4]。
目前QNs的检测方法主要为高效液相色谱[5-10]和高效液相色谱-质谱法[11-17]。高效液相色谱法的灵敏度较低,分析时间长,存在目标物保留时间随基质不同而漂移进而影响定性等问题;高效液相色谱-质谱法分析速度快、灵敏度高、选择性和特异性好,是目前药物残留方面较为先进的检测技术。样品的前处理方法对于药物的检测也至关重要。QNs的前处理方法一般为酸化乙腈提取,正己烷液-液萃取除脂,旋蒸浓缩后复溶定容。这种前处理存在的问题有两点:耗费有机溶剂多,耗时长;因不同种类水产品含有一定水分,旋蒸时不易蒸干,影响定容,导致回收率低和重现性差,甚至会影响定性。QuEChERS是由美国化学家Lehotay和德国的Anastassiadas于2003年提出的一种快速、简单、便宜、有效、可靠和安全的样品前处理技术[18],在提取液中直接加入吸附剂粉末进行净化,即分散固相萃取,最初应用于农药残留中[18-19]。近年来已越来越多的应用于各种抗生素和激素的前处理技术中[20-24]。而将QuEChERS技术作为水产品和食品中QNs检测的前处理方法并不多见[25],已有的方法也较为繁琐。本实验经过比较研究,采用了颇为实用和快捷的分散固相萃取技术,结合超高效液相色谱-串联质谱,建立了一种同时检测水产品中14 种QNs残留的方法,14 种药物在6 min内即可完全出峰。本方法简便快速,消耗有机溶剂少,定量限和回收率均满足已有的最高残留限量标准,适合水产品中多种QNs的确证和定量测定。
1 材料与方法
1.1 材料与试剂
14 种标准品分别为氟甲喹(flumequine,FLU)、萘啶酸(nalidixic acid, NAL)、恶喹酸(oxolinic acid,OXO)、环丙沙星(ciprofloxacin,CIP)、单诺沙星(danofloxacin,DAN)、双氟沙星(difloxacin,DIF)、恩诺沙星(enrofloxacin,ENR)、麻保沙星(marbofloxacin,MAR)、诺氟沙星(norfloxacin,NOR)、沙拉沙星(sarafloxacin,SAR)、氧氟沙星(ofloxacin,OFL)、洛美沙星(lomefloxacin,LOM)、司帕沙星(sparfloxacin,SPA)、培氟沙星(pefloxacin,PEF)(纯度98.5%~99.0%) 德国Dr. Ehrenstorfer公司;乙腈、甲醇、甲酸、正己烷均为色谱纯;碳酸钠、氯化钠、硫酸镁均为分析纯;N-丙基乙二胺(N-propylethylendiamine,PSA)吸附剂、十八烷基键合硅胶吸附剂(C18) 美国Agilent公司;实验用水为Milli-Q超纯水。
标准溶液的配制:分别准确称取14 种标准品0.010 g(有盐酸盐结合物的标准品按照分子式换算成相应克数),加入200 μL甲酸,用乙腈溶解稀释并定容至100 mL(若加入乙腈不溶解,则加入100 μL纯水),配制成100.0 μg/mL的标准储备液,使用时用初始流动相稀释成所需质量浓度。
1.2 仪器与设备
AcquityTM型超高效液相色谱仪、Quattro PremierTMXE型四极杆质谱仪、0.22 μm过滤膜 美国Waters公司;T18匀浆机、MS2漩涡混合器 德国IKA公司;Centrifuge 5810高速离心机 德国Eppendorf公司;N-EVAP112氮吹仪 美国Organomation公司;R-215旋转蒸发仪 瑞士Büchi公司;Vi siprepTMDL固相萃取装置美国Supelco公司。
1.3 方法
1.3.1 样品前处理
实验所用的水产品为草鱼、对虾和大黄鱼,按照SC/T 3016—2004《水产品抽样方法》的规定处理后于-18℃密封保存。使用前先解冻。
称取(5±0.02) g处理后的样品于50 mL 离心管中,加入20 mL 5%甲酸-乙腈溶液,涡旋提取3 min,再加入5.0 g 无水硫酸钠和2.0 g氯化钠,继续涡旋混匀1 min,6 000 r /min离心5 min,准确移取10 mL于15 mL离心管中,依次加入200 mg C18、200 mg PSA和500 mg无水硫酸镁,轻微涡旋振荡1 min,6 000 r /min 离心5 min,准确移取5.0mL上清液于10 mL 离心管中,45 ℃水浴下氮气吹干,加入1 mL 0.2%甲酸-甲醇(9∶1,V/V)的定容液,漩涡混匀,经0.22 μm滤膜过滤后供超高效液相色谱-串联质谱法测定。
1.3.2 色谱条件
ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm);柱温40 ℃;进样量10 μL;流动相:A为 0.2%甲酸,B为甲醇,梯度洗脱:0~4 min,10%~40% B;4~5 min,40%~10% B;5.0~6.5 min,B保持10%不变;流量0.3 mL/min。
1.3.3 质谱条件
离子源:电喷雾离子源(electrospray ionization,ESI);扫描方式:正离子扫描;检测方式:多反应监测(multiple reaction monitoring,MRM);毛细管电压:3.5 kV;离子源温度:120 ℃;脱溶剂气温度:380 ℃;锥孔气为高纯氮气;流量:50 L/h;脱溶剂气为高纯氮气,流量:600 L/h。
2 结果与分析
2.1 色谱条件的优化
使用2.0 μg/L的混合标准溶液,考察液相色谱流动相类型和酸度对灵敏度和分离度的影响。选择甲醇、乙腈以及甲醇和乙腈的混合溶液分别作为流动相的有机相部分。实验表明,甲醇作为流动相时各目标物的出峰时间比乙腈延后1~2 min,峰强度增强,峰形尖锐;采用甲醇和乙腈混合溶液比使用甲醇时各物质的峰强度略有降低,保留时间相差不大,为实现峰形尖锐、灵敏度高、操作简便,综合考虑后选择甲醇作为有机相。流动相中加入一定的有机酸可显著提高两性QNs的分离度和离子化效率,而对于C-7环上缺少哌嗪环的酸性QNs的影响不大。分别采用0.1%、0.2%和0.3%的甲酸溶液作为流动相,比较14 种药物的峰形和灵敏度,最终选择0.2%的甲酸溶液作为流动相。如图1所示,当甲酸体积分数超过0.2%时多数目标物的灵敏度显著下降,此时过多有机酸的加入已经抑制了喹诺酮类药物的电离。

图1 不同体积分数甲酸溶液条件下14 种喹诺酮药物的峰高大小Fig.1 The peak intensity of 14 QNs at different concentrations of formic acid
2.2 质谱条件的优化
喹诺酮类化合物分子结构中含有羰基等多电子基团,易于在ESI源的正离子模式下形成[M+H]+准分子离子。在ESI+模式下分别对1.0 μg/mL的14 种药物的单标溶液进行一级质谱全扫描分析,得到每种药物的分子离子,比较不同锥孔电压下的母离子强度,确定每个母离子有最大响应值时的最佳锥孔电压值。在最佳锥孔电压下,通过子离子扫描确定14 种药物的碎片离子,并选择响应强度较高的两个子离子作为定量离子和定性离子,同时对碰撞电压进行优化,确定碎片离子有最大响应强度时的最佳碰撞电压值。14 种喹诺酮药物的质谱优化参数见表1。
2.3 前处理的优化
C18、PSA、石墨化炭黑(graphitized carbon black,GCB)是QuEChERS中常用的吸附剂。C18除脂效果好;PSA可去除脂肪酸、糖类、酚类和极性色素;GCB可有效去除色素。水产品中蛋白质和脂肪含量高,虾中色素含量高,考虑到GCB易吸附像QNs一类的带有苯环官能团的化合物,因此选择PSA和C18作为净化剂。分别在相同的加标复溶液中添加100 mg的PSA和C18,涡旋混匀并离心后上机测定QNs的含量,将峰面积与未加吸附剂复溶液的QNs峰面积比较,以考察这两种吸附剂对QNs的吸附和净化情况。结果表明100 mg的C18和PSA对14 种喹诺酮的吸附率分别低于8%和12%,但净化效果不理想。根据提取液中脂肪和水分含量特点,以吸附剂用量少和净化效果好为原则,进一步比较了200~300 mg C18、200~300 mg PSA、500 mg和1 000 mg无水硫酸镁按不同比例组合的净化效果和回收率。C18为200 mg时的净化效果明显优于100 mg时,增加到300 mg时净化效果无明显变化;PSA为300 mg时的净化效果明显高于100 mg和200 mg时,但回收率有明显降低。添加不同组合净化剂的14 种QNs的加标回收率情况见图2,最终选择200 mg PSA、200 mg C18和500 mg无水硫酸镁为组合净化剂。

图2 14 种喹诺酮类药物添加不同组合净化剂的回收率情况Fig.2 The recoveries of 14 QNs under different combinations of purifying agents
2.4 基质效应及消除
质谱法普遍存在基质效应,尤其是对于动物源等基质复杂的样品,影响测定结果精密度和准确度。因此本研究分别用初始流动相和空白基质溶液配制标准曲线,考察了基质效应的影响。结果表明,当采用流动相直接配制标准曲线时,14 种喹诺酮类药物普遍存在程度不一的基质减弱效应,如图3所示。实际检测中采用空白基质溶液配制曲线可有效减轻基质干扰,使定量分析更加准确。

图3 基质效应消除前后14种药物的回收率Fig.3 Effects of different purification conditions on the recoveries of 14 QNs
2.5 线性范围、检出限与定量限
将6 个水产品空白样品按上述前处理方法提取和净化,每个样品均用初始流动相定容至1 mL,获得6 mL空白基质溶液。用此溶液将混合标准溶液稀释成0.5、2.5、5.0、10.0、20.0、50.0 μg/L的系列溶液,在2.3节所示的条件下上机测定。以目标物定量离子的峰面积为纵坐标,质量浓度为横坐标得到线性回归方程,14 种药物的线性相关系数均大于0.995,表明在0.5~50.0 μg/L范围内各化合物均呈良好的线性关系。逐级降低基质样品加标量,最终分别将3倍信噪比和10倍信噪比确定为方法检出限和定量限,结果见表2。14 种药物的检出限为0.40~1.00 μg/kg,定量限为1.00~3.00 μg/kg。5.0、15.0 μg/kg三个水平的14 种QNs的混合标准溶液,每个水平平行测定5 次,计算回收率和精密度。日间精密度采用5.0 μg/kg 添加水平连续测定5 d,计算日间精密度。大黄鱼空白加标样品中14 种QNs的MRM谱图见图4。回收率和精密度结果见表3。由表3可知,14 种QNs药物在3个加标水平下的平均回收率为82%~90%,日内精密度和日间精密度均小于11.0%,表明该方法的准确性和重复性良好。
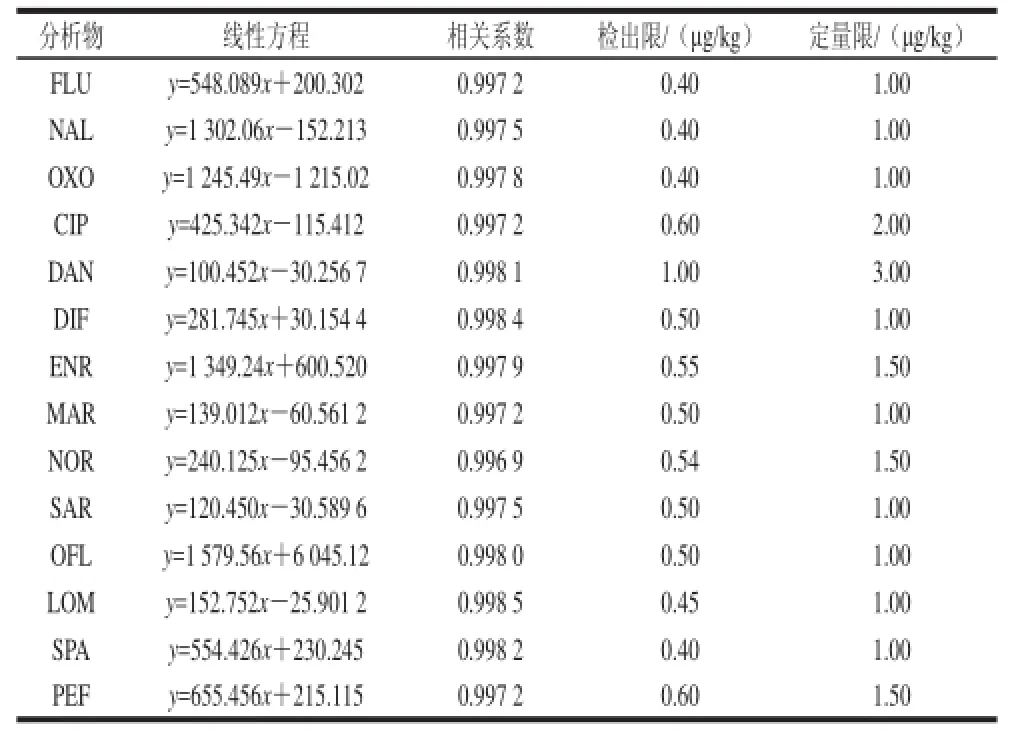
表2 14 种喹诺酮类药物的线性关系、检出限及定量限Table 2 Regression equations, limits of detection, limits of quantity of 14 quinolone antibiotics

2.6 方法回收率和精密度
分别在对虾、草鱼和大黄鱼空白基质中添加1.0、
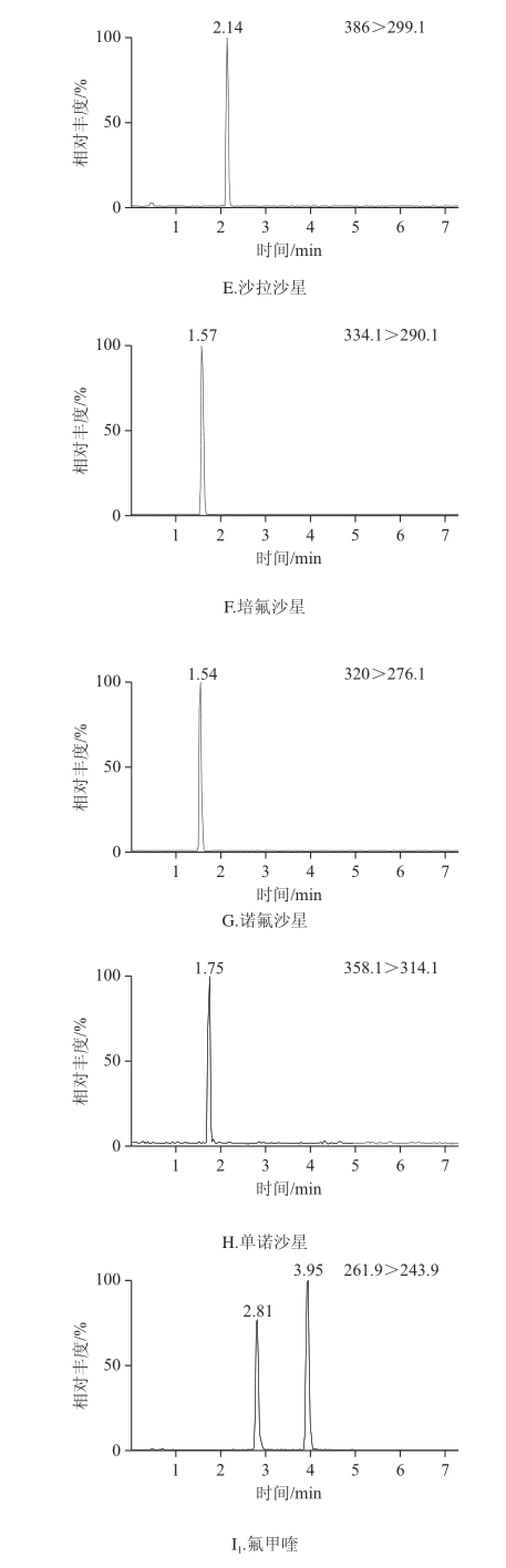


图4 5.0 μg/kg的大黄鱼空白加标样品中14 种QNs药物MRM谱图Fig.4 MRM chromatograms of 14 QNs in Pseudosciaena crocea spiked at 5.0 μg/kg

表3 大黄鱼样品中添加回收率及方法的精密度实验( =5)Table 3 Recovery and precision (RSD) of 14 QNs from fortified Pseudosciaena crocea samples ( = 5)
2.7 实际样品测定
从市场上购买不同种类的海产品,利用建立好的方法对14 种QNs类药物进行测定,结果均未检出14 种喹诺酮类药物。利用建立好的方法参加了英国分析实验室能力验证国际比对项目中水产品中10 种喹诺酮类药物检测,共检测出3 种加标样品,且结果在给定值的允许范围内,表明本方法可以作为QNs类药物的常规检测。
3 结 论
本实验建立了分散固相萃取-超高效液相色谱-串联质谱法测定水产品中14 种QNs类药物的方法,样品前处理简单,易于操作,采用超高效液相-质谱检测,显著缩短了色谱分析时间,结果灵敏度高,准确可靠。
[1] 张玉岩, 许立春. 喹诺酮类药物的合理利用[J]. 吉林医药, 2008, 29(5): 755-756.
[2] 李俊锁, 邱月明, 王超. 兽药残留分析[M]. 上海: 上海科学技术出版社, 2002: 256-299.
[3] 周启星, 罗义, 王美娥. 抗生素的环境残留、生态毒性及抗性基因污染[J]. 生态毒理学报, 2007, 2(3): 243-251.
[4] 李锋格, 苏敏, 李晓岩. 分散固相萃取-超高效液相色谱-串联质谱法测定鸡肝中磺胺类、喹诺酮类和苯并咪唑类药物及其代谢物的残留量[J]. 色谱, 2011, 29(2): 120-125.
[5] 赵思俊, 李存, 江海洋, 等. 高效液相色谱检测动物肌肉组织中7 种喹诺酮类药物的残留[J]. 分析化学, 2007, 35(6): 786-790.
[6] 钱卓真, 苏秀华, 魏博娟, 等. 高效液相色谱法同时测定水产品中6种喹诺酮药物的残留[J]. 食品科学, 2010, 31(6): 185-189.
[7] 周萍, 胡福良, 徐权华, 等. 高效液相色谱-荧光检测器测定蜂王浆中氟喹诺酮类药物残留的研究[J]. 食品科学, 2009, 30(6): 222-225.
[8] MOEMA D, NINDI M M, DUBE S. Development of a dispersive liquid-liquid microextraction method for the determination of fluoroquinolones in chicken liver by high performance liquid chromatography[J]. Analytica Chimica Acta, 2012, 730(2): 80-86.
[9] SCHNEIDER M J, BRADEN S E, REYES-HERRERA I, et a1. Simultaneous determination of fluoroquinolones and tetracyclines in chicken muscle using HPLC with fluorescence detection[J]. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Science, 2007, 846(1/2): 8-13.
[10] EVAGGELOPOULOU E N, SAMANIDOU V F. HPLC confirmatory method development for the determination of seven quinolones in salmon tissue (Salmo salar L.) validated according to the European Union Decision 2002/657/EC[J]. Food Chemistry, 2013, 136(2): 479-484.
[11] 王志杰, 冷凯良, 孙伟红, 等. 高效液相色谱-串联质谱法同时测定鳗鱼和虾中残留的33 种喹诺酮和磺胺类药物[J]. 色谱, 2009, 27(2): 138-143.
[12] 刘芃岩, 申杰, 刘磊. 复合模板印迹聚合物净化液相色谱-质谱联用法测定鱼肉中氟喹诺酮类残留[J]. 分析化学, 2012, 40(5): 693-698.
[13] 李雅丽, 郝晓蕾, 冀宝庆, 等. HPLC-ESI-MS/MS测定动物性食品中19 种喹诺酮类药物残留的研究[J]. 食品科学, 2008, 29(8): 502-506.
[14] 马建民, 夏曦, 李晓薇, 等. 阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13 种喹诺酮类药物[J]. 中国食品卫生杂志, 2012, 25(3): 249-253.
[15] SMITH S, GIESEKER C, REIMSCHUESSEL R, et al. Simultaneous screening and confirmation of multiple classes of drug residues in fish by liquid chromatography-ion trap mass spectrometry[J]. Journal of Chromatography A, 2009, 1216(46): 8224-8232.
[16] BAILAC S, BARRON D, BARBOSA J. New extraction procedure to improve the determination of quinolones in poultry muscle by liquid chromatography with ultraviolet and mass spectrometric detection[J]. Analytica Chimica Acta, 2006, 580(2): 163-169.
[17] GAJDA A, POSYNIAK A, ZMUDZKI J, et al. Determination of ( uoro) quinolones in eggs by liquid chromatography with uorescence detection and con rmation by liquid chromatographytandem mass spectrometry[J]. Food Chemistry, 2012, 135(2): 430-439.
[18] ANASTASSIADES M, LEHOTAY S J, STAJNBAHER D, et al. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce[J]. Journal of AOAC International, 2003, 86(2): 412-431.
[19] 黄宝勇, 潘灿平, 张微, 等. 应用分析保护剂补偿基质效应与气相色谱-质谱快速检测果蔬中农药多残留[J]. 分析测试学报, 2006, 25(3): 11-16.
[20] 董静, 宫小明, 张立, 等. QuEChERS-高效液相色谱法同时检测动物组织中的克球酚、地克珠利和磺胺类药物残留量[J]. 中国卫生检验杂志, 2008, 18(1): 26-29.
[21] 王炼, 黎源倩, 王海波, 等. 基质固相分散超高效液相色谱串联质谱法同时测定畜禽肉和牛奶中20 种兽药残留[J]. 分析化学, 2011, 39(2): 203-207.
[22] 王重洋, 王远鹏, 王宁, 等. 基质固相分散-超快速液相色谱法测定牛肉中磺胺类兽药[J]. 分析化学, 2013, 41(1): 83-87.
[23] 罗辉泰, 黄晓兰, 吴惠勤, 等. QuEChERS/液相色谱-串联质谱法同时测定鱼肉中30 种激素类及氯霉素类药物残留[J]. 分析测试学报2011, 30(12): 1329-1337.
[24] AGUILERA-LUIZ M M, MARTNEZ-VIDAL J L, ROMEROGONZLEZ R, et al. Multi-residue determination of veterinary drugs in milk by ultra-high-pressure liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography A, 2008, 1205(1/2): 10-16.
[25] 曹军, 陈勇, 杨瑞章, 等. 分散固相萃取-高效液相色谱-串联质谱法测定水产品中19 种喹诺酮类兽药残留[J]. 中国兽药杂志, 2011, 45(7): 21-25.
Determination of 14 Quinolone Antibiotics in Aquatic Products by Dispersive Solid-phase Extraction and Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry
LI Pei-pei, ZHANG Xiao-jun, MEI Guang-ming, YAN Zhong-yong, LONG Ju, LIU Qin, GUO Yuan-ming*
(Zhejiang Province Key Laboratory of Mariculture and Enhancement, Marine Fisheries Research Institute of Zhejiang, Zhoushan 316100, China)
A method for the simultaneous analysis of 14 quinolone (QN) residues in aquatic products by dispersive solidphase extraction and liquid chromatography-tandem mass spectrometry (UPLC-MS-MS) was established. The sample was extracted with acetonitrile (containing 5% formic acid) and followed by salting-out. Clean up of the extracts was performed using PSA and C18during the dispersive solid-phase extraction proced ure. After evaporated to dryness under a stream of nitrogen, the analyte s were then separated on a Waters ACQUITY UPLCTMBEH C18column (50 mm × 2.1 mm, 1.7 μm) using binary mobile phase gradient with water containing 0.2% formic acid and methanol. The targeted compounds were detected under a multiple reaction monitoring (MRM) mode and quantified by an external standard method. The linearity of all 14 QNs in the range from 0.50 to 20.0 μg/L exhibited correlation coefficients greater than 0.995. The limits of detection (LOD) and the limits of quantification (LOQ) were 0.4–1.0 and 1.0–3.0 μg/kg, respectively. The average recoveries of 14 QNs at three level spiked concentrations ranged from 82% to 90%, with relative standard deviations (n = 5) less than 11.0%. This method was simple, rapid, sensitive and reliable, and could be applied to determine 14 QNs in aquatic products.
dispersive solid-phase extraction; ultra performance liquid chromatography-tandem mass spectrometry (UPLCMS-MS); aquatic products; quinolones
O657.63
A
1002-6630(2014)24-0265-06
10.7506/spkx1002-6630-201424051
2014-03-12
浙江省重点科技创新团队项目(2010R50028);浙江省科技计划项目(2013C37072;2012F20026)
李佩佩(1986—),女,工程师,硕士,研究方向为渔业环境监测和水产品质量安全。E-mail:liwanzhao999@163.com
*通信作者:郭远明(1977—),男,高级工程师,硕士,研究方向为渔业环境和水产品质量安全调查、检测和评价。E-mail:guoyuanming@msn.com
