基于454 测序的碧桃花瓣组织转录组核苷酸变异1)
2014-03-06陈赢男戴晓港马秋月刘海琳李淑娴
陈赢男 戴晓港 马秋月 刘海琳 李淑娴
(南京林业大学,南京,210037)
单核苷酸多态性(single nucleotide polymorphism,SNP)是许多物种基因组中最常见的变异形式,具有数量多、分布广、突变率低、可稳定遗传等特点,被认为是最具发展潜力的分子标记[1-2],在遗传图谱构建、数量性状定位、分子标记辅助育种等方面有着广泛的应用[3-5]。如,Hyten 等[6]利用1,536 个SNP 标记构建了一个高密度的大豆数量性状遗传图;Yu 等[7]利用高密度的SNPs 图谱成功检测到与稻米粒型粒重相关的数量性状位点。此外,某些位于基因内部的SNP 位点可以直接影响基因的功能[8-9]。Fan 等[10]报道了水稻GS3 基因第二外显子上C-A 单核苷酸突变导致长粒品种中GS3 蛋白翻译提前终止。与SNP 相比,插入缺失长度多态性(insertion deletion length polymorphism,InDel)是基因组中另一较为丰富的多态性资源[11],InDel 所产生的序列改变更为显著,故其对基因功能的影响也更大[12]。
随着测序技术的发展,转录组测序已成为解读基因组功能元件、揭示基因表达与表型之间内在联系的重要手段,同时也成为检测分子标记的重要数据源。基于转录组及其它EST 序列开发的分子标记已被广泛运用于大豆(Giycine max)、玉米(Zea mays)、杨树(Populus deltoides)等[13-15]等多个物种的遗传图谱构建和遗传多样性分析中。
碧桃(Prunus persica)是蔷薇科桃属的观花木本植物,具有花型丰富、花色艳丽、花香独特等特点,已被广泛应用于园林绿化。随着桃树[16]基因组测序工作的完成,越来越多转录组信息的公布[17-18],桃树分子标记的开发也日益受到重视[18]。如曹珂等[19]以桃“红垂枝”与“白花山碧桃”为亲本,构建了一个包含206 个标记(18 个SSR,126 个AFLP、61个SRAP 和1 个形态学标记)遗传图谱,并将雌蕊发育和单瓣/重瓣性状进行定位;Verde 等[20]对56 个桃树品种进行重测序,检测到1 022 354 个SNP。为了进一步丰富碧桃分子标记类型及数量,本研究利用454 高通量测序技术对碧桃花瓣组织转录组进行测序,并对获得的数据进行SNP/InDel 位点查找和分析。
1 材料与方法
桃花转录组测序材料选自南京市情侣园内一株树龄约5 a 的碧桃(Prunus persica L.)。于2013年3月盛花期采集花瓣,并用液氮迅速冷冻,置于冰箱中-80 ℃保存备用。
RNA 提取与cDNA 合成:采用CTAB 法[21]提取总RNA,样品中残留的基因组DNA 利用DNase I(RNase Free)来去除,参照Takara 公司的使用说明进行操作。采用Oligotex mRNA 纯化试剂盒(Qiagen)对mRNA 进行纯化,然后使用cDNA Synthesis System Kit(Roche)合成cDNA。
cDNA 文库构建与转录组测序:使用Rapid Library Prep Kit(Roche)构建测序文库,并用Agilent 2100 Bioanalyzer 对文库质量进行检测。参照标准的测序试验流程(Roche),利用454 GS FLX 测序仪(454 Life Sciences)对构建好的cDNA 文库进行测序,利用软件Newbler v2.8 software(Roche)进行序列拼装。
核苷酸变异分析:使用拼接后的序列作为参考序列,利用软件GS Reference Mapper (Roche Inc.,version 2.8)将原始测序片段与参考序列进行比对,检测核苷酸变异位点,包括转换、颠换、插入和缺失。软件参数设置及序列筛选标准参考Ma 等[22]的研究方法。
含SNP/InDel 序列的功能注释:利用Blast2go(http://www.blast2go.org)程序,对含有SNP/In-Del 的重叠群序列进行功能注释(E <10-5)并获取GO 编号。利用BGI WEGO(http://wego.genomics.org.cn/cgi-bin/wego/index.pl)将获得的GO 编号进行功能分类。
2 结果与分析
2.1 转录组测序及序列组装
采用454 GS FLX 测序平台对碧桃花瓣组织转录组测序共获得1 556 684 条序列,平均读长446 bp,共有695.34 Mb 的数据量。去除低质量序列后有1 492 289 条序列参与组装,共得到22 762 个重叠群(≥100 bp)和72 719 个singleton。在所得到的重叠群中,长度大于等于500 bp 的重叠群有14 006 个(61.53%),最大重叠群为14 330 bp,重叠群N50 为1 426 bp,平均重叠群(≥500 bp)大小为1 273 bp。数据拼接结果见网址:http://115.29.234.170/。
2.2 SNP 位点查找
利用454 newbler GS Mapper 软件,将原始序列比对到组装好的转录组参考序列上,在2 561 个重叠群中共检测到9 836 个SNP 位点,平均每2 013 bp就有一个SNP。SNP 位点中,转换6 152 个(A/G、C/T),颠换3 684 个(A/C、A/T、G/T、G/C)。其中,A/G 突变最多,占总数的31.83%,C/T 突变其次,占到30.71%;C/G 突变最少,占总数的8.42%(表1)。统计结果显示,本实验所检测到的SNP 大部分集中于长度在100 ~2 100 bp 范围内的重叠群上,只有1867 个(18.97%)SNP 位点在长度大于2 100 bp的重叠群上检测到,这是由于组装后得到的大于2 100 bp 的重叠群本身就比较少。所以本研究检测到的SNP 数据库见网址:http://115.29.234.170/。
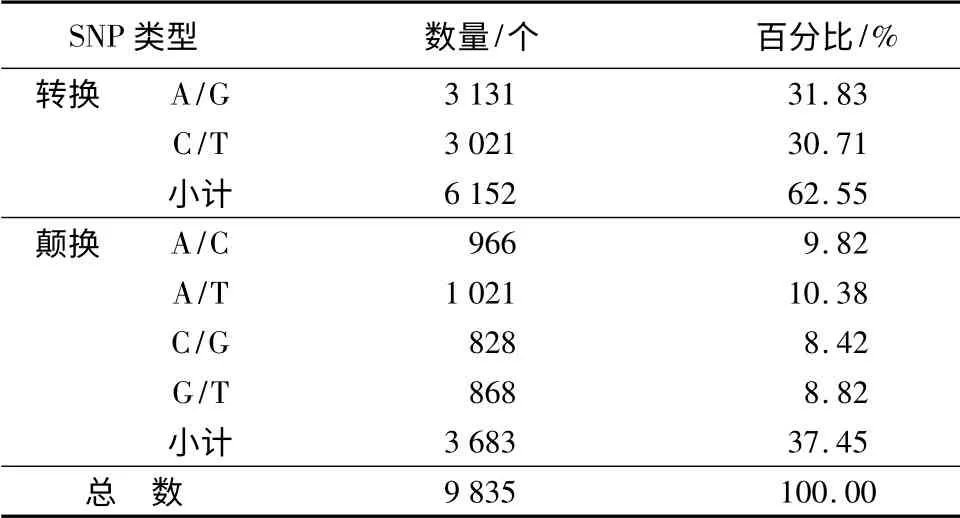
表1 SNP 分类型统计分析
2.3 InDel 位点查找
在933 个重叠群中检测共出1 550 个InDel 位点,包括1,257 个插入突变和293 个缺失突变。与SNP 位点相比,InDel 位点占所有核苷酸变异位点的13.61%。核苷酸插入和缺失突变的变化范围分别在1 ~53 bp 和1 ~47 bp,其中单核苷酸插入、缺失占所有InDel 位点的34.71%,小于或等于10 bp 的In-Del 总数占InDel 总量的83.87%。在插入突变中,有468 个位点为单核苷酸插入;在缺失突变中,检测到最多的是三核苷酸缺失(78 个),其次为单核苷酸缺失(70 个)。无论是插入还是缺失突变,都基本上呈现出位点数目随核苷酸变异长度增加而减少的趋势(表2)。
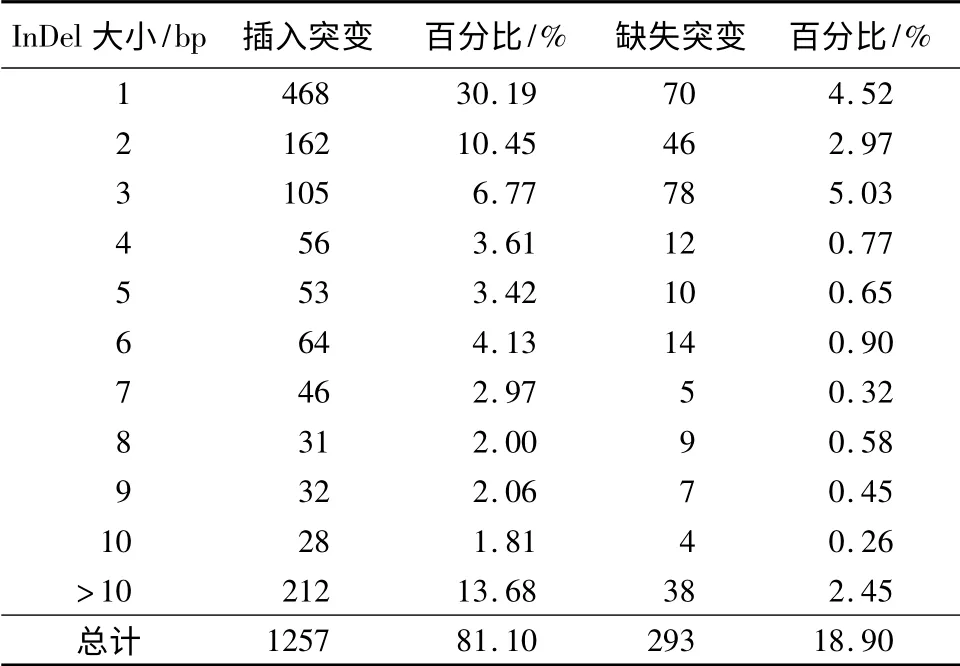
表2 InDel 位点数量随其长度变化分布
2.4 含SNP/InDel 重叠群序列的功能
以NCBI nr 数据库基因注释为参考,将3 159 个含有SNP/InDel 的重叠群序列进行BLAST 比对,共有2 655 条(84.05%)序列被匹配,共有8 417 个SNP 位点匹配到2 655 个注释基因上。在成功注释的重叠群中有2 204 个被GO 分类,功能分析发现有1 300 个重叠群参与分子功能、490 个重叠群参与细胞学组件、681 个重叠群参与生物学途径(图1)。
3 结论与讨论
分子标记的快速发展为观赏园艺植物研究领域开辟了新的途径。近年来,分子标记技术在花卉品种鉴别、遗传图谱构建、分子标记辅助育种等方面的应用也日益广泛[23-25]。桃属植物因其基因组较小(2c = 265 Mb,2n = 16)被视为蔷薇科的模式植物[26],桃全基因组测序工作的完成使得大规模分子标记的开发成为可能。本文通过对碧桃花瓣组织转录组测序,拼接后共得到22 762 个重叠群(≥100 bp),其中2 561 个重叠群上检测到9 836 个SNP 位点,平均每2 013 bp 有一个SNP,发生转换和颠换概率的比值(Ts/Tv)为1.67,大于理论上0.5 的比值[27]。对于实际中Ts/Tv值大于0.5 的现象(又称“转换偏差”),一种假说认为是由于一些内在的特征如化学组成、结构差异(嘌呤和嘧啶)[28];一种认为这可能由于进化选择导致,因为转换偏差更易于保持所编码蛋白的原有结构,降低有害突变的发生[29-30]。
在多数有关SNP 变异类型的报道中,C/T 所占比例最高,目前人们普遍认同的观点是CG 二联码的C 常被甲基化,容易脱氨基转换成T 从而成为突变热点[31-32]。在碧桃花瓣转录组转换位点中,A/G、C/T 两种变异类型几乎相等C/T(1.04:1),但C/T 所占比例与大麦、小麦、辣椒等物种中的SNP 碱基变异类型相似[3,33-34]。在颠换变异类型中,A/T 发生的频率最高,C/G 发生的频率最小,部分原因可能是由于桃树基因组本身GC 含量就比较小(37.07%)导致的。
遍布于整个基因组的InDel 频率仅次于SNP[35],InDel 作为重要的遗传标记已被广泛用于作物连锁图谱的构建及多样性研究[36]。本研究在933 个重叠群中检测出1 550 个InDel 位点,1 bp 长InDel 的比例超过InDel 总量的30%,插入和缺失的数量都随着InDel 长度的增加而下降。这种长度与数量的相关性在许多植物中都有报道[22,28,37]。较长的InDel 可能对生物有不同程度的危害,其比例也相对很少[38]。
随着碧桃基因组、转录组测序研究的不断深入,其SNP/InDel 资源将更加丰富。本研究中检测到SNP/InDel 位点将有助于研究核苷酸变异引起的蛋白质功能的变化,同时,这些多态性位点可以开发成分子标记应用到基因的图位克隆及分子标记辅助育种中。
[1] Brooker A J.The essence of SNPs[J].Gene,1999,234(2):177-186.
[2] Gray I C,Campbell D A,Spurr N K:Single nucleotide polymorphisms as tools in human genetics[J].Human Molecular Genetics,2000,9(16):2403-2408.
[3] Chao S,Zhang W,Akhunov E,et al.Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.)cultivars[J].Molecular Breeding,2008,23(1):23-33.
[4] Niu P X,Huang Z,Li C C,et al.Cloning,chromosomal localization,SNP detection and association analysis of the porcine IRS-1 gene[J].Molecular Biology Reports,2009,36(8):2087-2092.
[5] Ren G,Chen H,Zhang L Z,et al.A coding SNP of LHX4 gene is associated with body weight and body length in bovine[J].Molecular Biology Reports,2010,37(1):417-422.
[6] Hyten D L,Choi I Y,Song Q,et al.A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping[J].Crop Science,2010,50(3):960-968.
[7] Yu H H,Xie W B,Wang J,et al.Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers[J].PLoS ONE,2011,6(3):e17595.
[8] Isshiki M,Morino K,Nakajima M,et al.A naturally occurring functional allele of the rice waxy locus has a GT to TT mutation at the 5’splice site of the first intron[J].The Plant Journal,1998,15(1):133-138.
[9] Abe F,Saito K,Miura K,et al.A single nucleotide polymorphism in the alternative oxidase gene among rice varieties differing in low temperature tolerance[J].FEBS Letters,2002,527(1):181-185.
[10] Fan C,Xing Y,Mao H,et al.GS3,a major QTL for grain length and weight and minor QTL for grain width and thickness in rice,encodes a putative transmembrane protein[J].Theoretical and Applied Genetics,2006,112(6):1164-1171.
[11] Edwards J D,Lee V M,McCouch S R.Sources and predictors of resolvable indel polymorphism assessed using rice as a model[J].Molecular Genetics and Genomics,2004,271(3):298-307.
[12] Petrov D A.Mutational equilibrium model of genome size evolution[J].Theoretical Population Biology,2002,61(4):531-544.
[13] Barbazuk W B,Emrich S J,Chen H D,et al.SNP discovery via 454 transcriptome sequencing[J].The Plant Journal,2007,51(5):910-918.
[14] 张新叶,宋丛文,张亚东,等.杨树EST-SSR 标记的开发[J].林业科学,2009,45(9):53-59.
[15] Shu Y,Li Y,Zhu Z,et al.SNPs discovery and CAPS marker conversion in soybean[J].Molecular Biology Reports,2011,38(3):1841-1846.
[16] Verde I,Abbott A G,Scalabrin S,et al.The high-quality draft genome of peach (Prunus persica)identifies unique patterns of genetic diversity,domestication and genome evolution[J].Nature Genetics,2013,45(5):487-494.
[17] Wang Lu,Zhao Shuang,Gu Chao,et al.Deep RNA-Seq uncovers the peach transcriptome landscape[J].Plant Molecular Biology,2013,83(4/5):365-377.
[18] 李雄伟,贾惠娟,高中山.桃基因组学及全基因组关联分析研究进展[J].遗传,2013,35(10):1167-1178.
[19] 曹珂,王力荣,朱更瑞,等.桃遗传图谱的构建及两个花性状的分子标记[J].园艺学报,2009,36(2):179-186.
[20] Verde I,Bassil N,Scalabrin S,et al.Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm[J].PLoS ONE,2012,7(4):e35668.
[21] Liu J,Yin T,Ye N,et al.Transcriptome analysis of the differentially expressed genes in the male and female shrub willows(Salix suchowensis)[J].PloS ONE,2013,8(4):e60181.
[22] Ma Q,Feng K,Yang W,et al.Identification and characterization of nucleotide variations in the genome of Ziziphus jujuba(Rhamnaceae)by next generation sequencing[J].Molecular Biology Reports,2014,41(5):3219-3223.
[23] 侯小改,尹伟伦,李嘉珏,等.部分牡丹品种遗传多样性的AFLP 分析[J].中国农业科学,2006,39(8):1709-1715.
[24] 田晔林,刘克锋,石爱平,等.一串红品种遗传多样性RAPD分析[J].中国农学通报,2006,22(5):76-78.
[25] 于恒秀,王淼,梁国华,等.ISSR 引物鉴定芍药栽培品种之间亲缘关系的初步研究[J].植物生理学通讯,2006,42(2):271-274.
[26] 徐勇.桃花发育相关MADS box 基因研究[D].北京:首都师范大学生命科学学院,2007.
[27] 赵辉,李启寨,李俊,等.相邻碱基组分与产生SNP 的转换或颠换在植物基因组中的研究[J].中国科学:C 辑,2006,36(1):1-8.
[28] 唐萍,王强,陈建群.茄科植物叶绿体基因组插入,缺失和核苷酸替代的发生方式及影响[J].遗传,2008,30(11):1506-1512.
[29] Li W H,Wu C I,Luo C C.Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications[J].Journal of Molecular Evolution,1984,21(1):58-71.
[30] Wakeley J.The excess of transitions among nucleotide substitutions:new methods of estimating transition bias underscore its significance[J].Trends in Ecology Evolution,1996,11(4):158-162.
[31] Bird A P.DNA methylation and the frequency of CpG in animal DNA[J].Nucleic Acids Research,1980,8(7):1499-1504.
[32] Gruenbaum Y,Naveh Many T,Cedar H,et al.Sequence specificity of methylation in higher plant DNA[J].Nature,1981,292:860-862.
[33] Sato K,Close T J,Bhat P,et al.Single nucleotide polymorphism mapping and alignment of recombinant chromosome substitution lines in barley[J].Plant and Cell Physiology,2011,52(5):728-737.
[34] 刘峰,谢玲玲,弭宝彬,等.辣椒转录组SNP 挖掘及多态性分析[J].园艺学报,2014,41(2):343-348.
[35] 孙宽,张素华,朱如心,等.新一代遗传标记:InDel 研究进展[J].法医学杂志,2013,29(2):134-139.
[36] 张体付,葛敏,韦玉才,等.玉米功能性Insertion/Deletion(In-Del)分子标记的挖掘及其在杂交种纯度鉴定中的应用[J].玉米科学,2012,20(2):64-68.
[37] Batley J,Barker G,O’Sullivan H,et al.Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data[J].Plant Physiology,2003,132(1):84-91.
[38] Tenaillon M I,Sawkins M C,Anderson L K,et al.Patterns of diversity and recombination along chromosome 1 of maize (Zea mays ssp.mays L.)[J].Genetics,2002,162(3):1401-1413.
