废茶活性炭的制备及其孔径结构的控制
2014-03-04宋磊张彬陈家元冯利
宋磊,张彬,陈家元,冯利
(1 华侨大学化工学院,福建 厦门 361021;2 云和县环境监测站,浙江 丽水 323600;3 河北工业职业技术学院环境与化学工程系,河北 石家庄 050091)
中国是世界上最大的产茶国之一,而福建省又是著名的产茶大省,每年都会加工大量的茶叶,因而伴随产生的废茶成为生活中最常见的废弃物之一。然而这些废弃物经合理的开发后仍具有很高的利用价值,如将其制备成护肤用品[1]、黏土砖[2]、除臭剂[3]等。2011年Panneerselvam等[4]还将废茶添加到磁性纳米颗粒中用以去除水溶液中的二价Ni,显示出独特的去除效果。
活性炭作为一种多孔性材料,因其丰富的比表面积以及良好的吸附性能而受到广泛的关注。尽管活性炭价格较为低廉,来源广泛,但由于其主要生产原料为不可再生矿物煤、木材等,这不仅增加了活性炭的生产成本,同时对生态环境的保护也不利。因此,低廉的废弃物作为制备活性炭的前体越来越得到人们的推广,不仅为活性炭的制备降低成本,还能为废弃物的治理提供更为可行的选择[5-7]。废茶本身具有较高的碳含量,近几年也开始有人将其制备成活性炭,均取得较好的效果。较早有见Yagmur等[8]采用微波辐照结合磷酸活化法将其制备成活性炭,BET达到1157m2/g,大连海洋大学的刘靖等[9]采用相近的方法制备废茶活性炭对亚甲基蓝吸附值达75.12mg/g 以上。传统的化学活化法制备的废茶活性炭比表面积也较高,Auta等[10]以乙酸钾为活化剂制备的废茶活性炭 BET最高达到 854.3m2/g,Gurten等[11]采用K2CO3为活化剂制备的废茶活性炭BET最高可达 1722m2/g,Gundogdu等[12]采用的ZnCl2活化法制备出BET达1066m2/g的废茶活性炭。Borah等[13]利用H3PO4活化制备的废茶活性炭BET最高可达 1285m2/g。Peng等[14]利用高温炭化及KOH活化法制备出超高比表面积的废茶活性炭,BET达2245~2841m2/g。由此可见将废茶制备成的活性炭具有较高的比表面结构,是一种良好的活性炭前体材料。
活性炭的孔径结构是影响其吸附性能的重要因素之一,因此控制活性炭的孔径结构对于提高其吸附性能有着重要的意义。然而,目前在活性炭制备过程中直接对活性炭孔径结构进行控制的研究还比较少,本文将通过选择不同的活化剂以及改变活化条件,以期制备出不同孔径结构的废茶活性炭。活性炭的活化方式主要分为物理活化法及化学活化法,物理活化包括水蒸气活化法和二氧化碳活化法等,有研究表明水蒸气活化相对于二氧化碳活化制备的活性炭烧失率低,比表面积大,微孔结构丰富[15]。相比而言,化学活化法制备的活性炭比表面高,孔径分布窄,活化剂包括ZnCl2、H3PO4、KOH、K2CO3等。其中ZnCl2活化法对于活性炭的微孔与中孔发展非常有利,Uçar等[16]由氯化锌活化制备石榴子活性炭,得到较高的微孔和中孔结构,国内的赵朔等[17]采用氯化锌活化制备出笋壳基中孔型活性炭。K2CO3是相对环保且常被用于食品添加剂的一种无毒无害物质,作为活化剂制备的活性炭均以微孔分布为主[18-19]。因而选取ZnCl2和K2CO3为活化剂对活性炭的微孔及中孔结构的控制具有一定的研究意义。此外,活性炭的制备条件包括活化剂的浓度、活化时间、活化温度等,对于活性炭的比表面积与孔径结构的影响较大[20]。因此本文主要以K2CO3和ZnCl2为活化剂制备废茶活性炭,通过改变活化温度、活化时间及活化剂浓度以控制活性炭的孔径结构,从而为活性炭孔径的控制及其吸附性能的提高提供理论依据。
1 实 验
1.1 活性炭制备
预处理:将收集到的福建铁观音废茶样用蒸馏水煮沸数次至滤出溶液无色,再于 105℃下烘干24h,压碎,置于干燥皿中保存备用。
将预处理后的废茶材料与活化剂(K2CO3、ZnCl2)以一定的质量比混合,加入适量的蒸馏水在玻璃容器中搅拌至充分混合后,置于鼓风烘箱中105℃烘干12h以上,得到浸渍料。
对所得到的浸渍料以 10℃/min的升温速率升至活化温度(350~900℃),活化0.5~2h,并以流量为 0.5L/m in的 N2为载气。活化后的样品用0.1mol/L盐酸煮沸,以除去残余的活化剂,并用热蒸馏水反复洗至滤液呈中性,并在 105℃下烘干,即得废茶活性炭。
对废茶活性炭(WTAC)以“WTAC-活化剂-活化温度-活化时间(活化剂与废茶的质量比值)”格式进行命名,如“WTAC-K-700-1(1)”表示K2CO3与废茶的质量比为1,在700℃下炭化1h制备而成的样品。此外,由废茶在700℃下水蒸气活化1h的样品标记为“WTAC-H-700-1”,由 800℃下在0.5L/minN2流量下直接炭化 1h的样品标记为“WTAC-800-1”。
废茶活性炭的产率见式(1)。
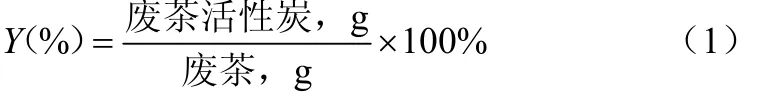
1.2 材料分析与表征
1.2.1 元素分析
采用EURO EA3000型有机元素分析仪有机元素分析仪测定废茶原料及所制备的废茶活性炭中的C、H、N、O含量(质量分数)。
1.2.2 热重分析
为检测材料的热分解特性,使用DTG-60(H)型岛津差热-热重分析装置,将废茶样品在氮气保护下,以10℃/m in的加热速率由室温加热至800℃。
1.2.3 比表面积及孔径分析
将材料在105℃下烘干12h以上,然后在200℃下进一步脱气 6h后,采用贝士德 3Ⅱ-2000PS1型比表面及孔径分析仪,于77K下测定氮气的吸附等温线。由BET多点法计算比表面积,BJH法计算中孔容及平均孔径,αs-plot法[21]计算总表面积、微孔容量、平均微孔径。
2 结果与讨论
2.1 原料分析
2.1.1 元素分析
废茶原料及废茶活性炭的元素成分如表1所示,废茶原料的 C、H、N、O等元素含量分别为48.34%、6.83%、6.08%、36.72%,与文献[8]报道的废茶元素成分相当,具有较高的碳含量。由表1可看出,经直接炭化的样品WTAC-800-1及K2CO3活化的样品WTAC-K-700-1(1)的C含量明显高于废茶原料。
2.1.2 热重分析
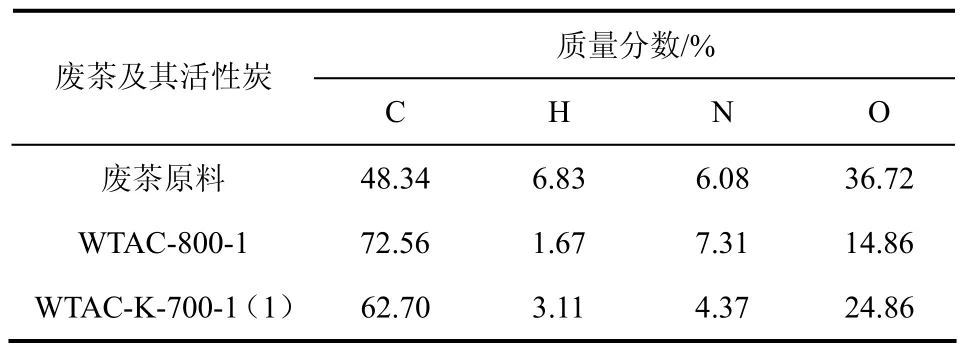
表1 废茶及废茶活性炭的元素成分分析
废茶(WT)热分析曲线如图1所示,在120℃之前主要是材料所吸附水分的失重过程,质量损失为4%左右。在200~400℃废茶材料的质量损失最为明显达到50%以上,此时主要是废茶主体材料的分解过程,包括木质素、纤维素、半纤维素等[11,22],与Yagmur等[8]的介绍相似,被认为是挥发性物质的演变过程。温度达到 500℃以后废茶材料的质量几乎不变,由此可见炭化过程已基本结束。
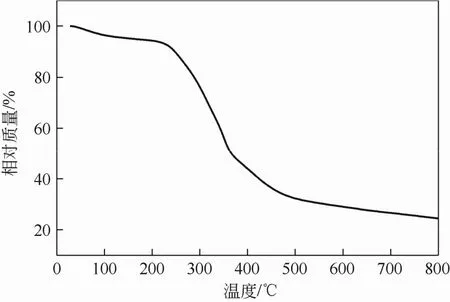
图1 废茶材料的热分析曲线
2.2 不同活化剂对废茶活性炭孔结构的影响
3 种不同活化方法制备的样品 WTAC-H-700-1、WTAC-Zn-700-1(1)、WTAC-K-700-1(1)的氮气吸附等温线如图2所示。3种材料的吸附等温线均呈现IUPAC中的Ⅰ型等温线,表明几种材料的孔结构中微孔占主导地位[23],其中,WTAC-K-700-1(1)的等温线在接近饱和蒸气压时,等温线又迅速上升,表现为 I-B型,说明WTAC-K-700-1(1)存在一定的大孔[24]。
3 种活化方式制备的样品产率、比表面及孔径结构如表2所示,K2CO3活化及水蒸气活化的产率均低于25%,而经ZnCl2活化后的样品产率相对较高,达43.88%,主要是由于ZnCl2作为一种Lew is酸在活化过程中促进材料中的易挥发组分芳构化、脱羟基、脱水、抑制焦油等,从而阻碍了挥发组分的释放,降低样品在活化过程中的质量损失[25]。
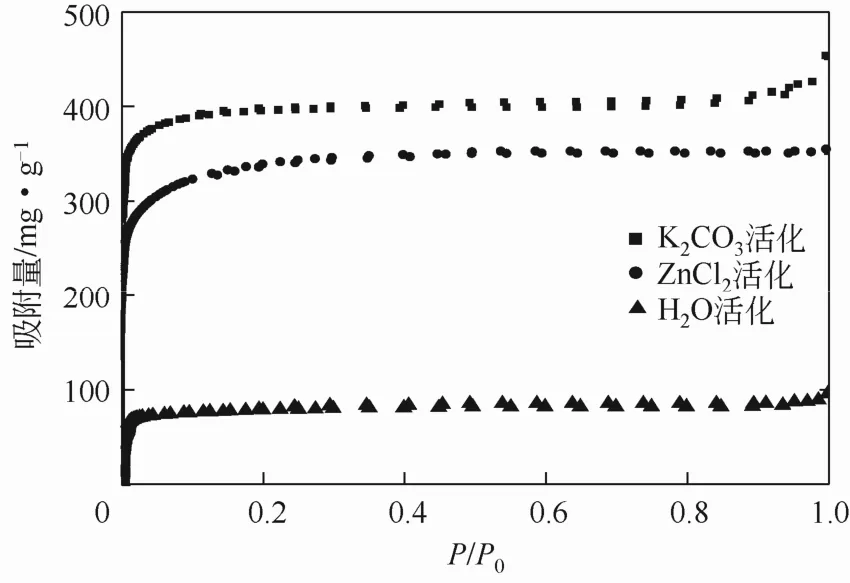
图2 不同活化剂下废茶活性炭的氮气吸附等温线
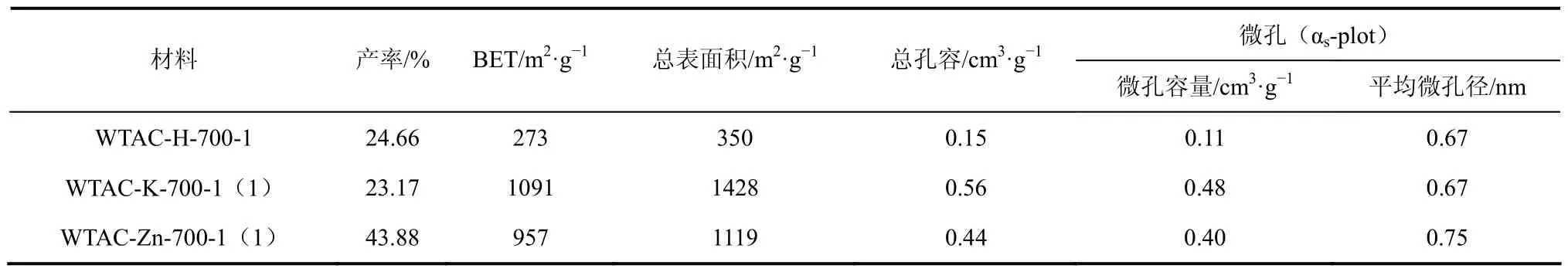
表2 不同活化剂对活性炭产率及孔结构的影响
通过各材料的比表面积可以看出经 K2CO3、ZnCl2活化制备的活性炭表面结构较发达,BET达1000m2/g左右,而水蒸气活化制备的WTAC-H-700-1的BET比表面积仅为273m2/g。这可能与活化剂的作用有关,在活化进程中,由于ZnCl2通过对纤维素内部分子结构的电解作用,使材料中的 H、O元素以水蒸气形式释放出去,从而形成多孔性结构[26-29];K2CO3在活化进程中由于高温作用,分解产生CO2和金属钾,K通过进入碳组织,增大碳原子层间的距离,而产生CO2作为活化剂和改性剂对碳材料进行了物理活化作用[30-31]。以上两个作用过程均为化学活化过程,水蒸气物理活化主要通过水分子的扩散然后在高温下与炭发生反应,从而促进孔隙的形成,这一过程对材料表面的造孔作用较弱。可见 K2CO3、ZnCl2化学活化法相比水蒸气物理活化法更有利于废茶活性炭多孔性结构的形成,BET达WTAC-H-700-1的3倍以上。
2.3 活化温度对废茶活性炭孔结构的控制
固定废茶与活化剂的浸渍比为1∶1,活化时间为1h,分别研究了K2CO3在500~900℃及ZnCl2在 350~700℃下的活化温度对活性炭产率及结构的影响。图3为活性炭产率随活化温度变化的趋势线,由于温度升高而形成的挥发性组分引起的脱水及消去反应使材料进一步失重,因而两种活性炭的产率随着活化温度的提高而逐渐降低。此外,由ZnCl2活化制备的废茶活性炭(WTAC-Zn)的产率显著高于 K2CO3活化制备的废茶活性炭(WTAC-K)。
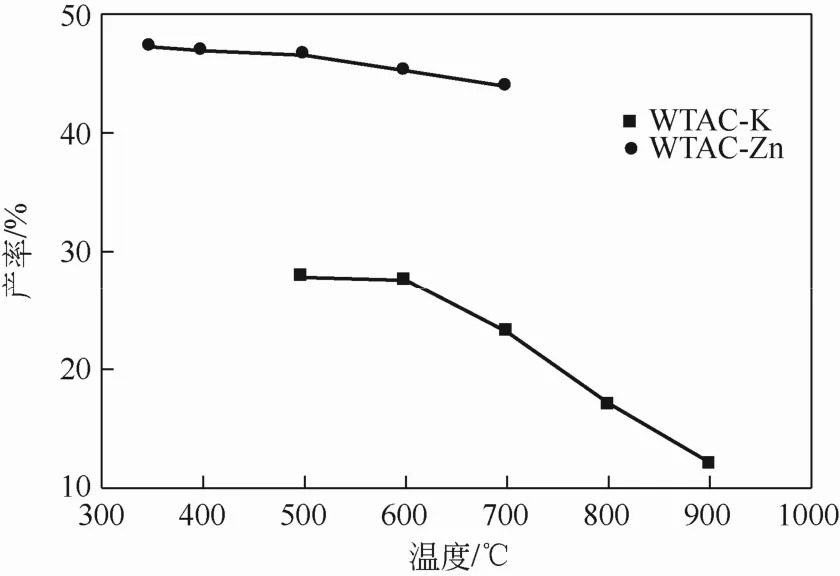
图3 WTAC产率随活化温度的变化趋势图
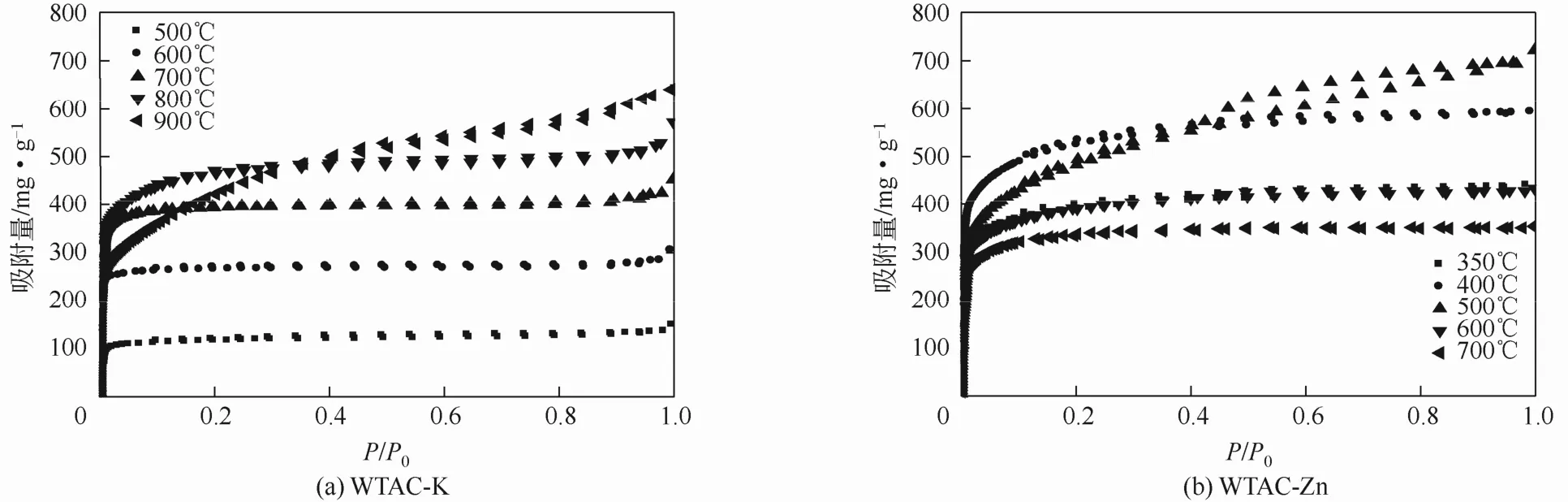
图4 不同活化温度下废茶活性炭的氮气吸附等温线
不同活化温度下制备的废茶活性炭氮气吸附等温线如图4所示。其中在 500~800℃下活化的WTAC-K呈现IUPAC中的Ⅰ-B型,表明它们的孔结构中微孔为主并伴有一定的大孔存在[24],当活化温度升至900℃,吸附等温线由Ⅰ-B型转变为Ⅳ型,可见WTAC-K产生了一定的中孔结构。在350~400℃时,WTAC-Zn的氮气吸附等温线呈Ⅰ-A型,当活化温度为 500℃时,氮气吸附等温线变为Ⅳ型并出现迟滞环,可见有中孔结构形成。继续升高活化温度,WTAC-Zn的氮气吸附等温线又变为Ⅰ-A型,中孔结构消失。
不同活化温度下制备的废茶活性炭的孔结构参数如表3所示,在500~800℃下WTAC-K的BET比表面积随温度的升高逐渐增大,当温度达到 900℃后BET比表面积反而减小,这主要是由于随温度的升高,活化剂在材料表面扩散并对碳孔壁的刻蚀作用加强,形成了孔结构及比表面扩展的交联结构,使材料的比表面积有所提高,继续升高温度则由于引起碳结构收缩,从而导致空隙度的减少,比表面积下降[32]。活化温度由 500℃升至700℃,材料的微孔容量随之增大,说明温度的升高促进了新的微孔结构形成,活化温度升到 800℃后活性炭的微孔容量及中孔容量均明显提高,说明在该温度下WTAC-K形成了新的微孔及中孔结构,这可能是由于 K2CO3经分解、还原作用生成的金属钾沸点为760℃,当活化温度超过该温度,钾蒸汽就会扩散入不同的碳层,促进新的孔结构[形成[30,33]。当活化温度达到 900℃后,微孔容量降低而中孔容量相对增大,平均微孔径由800℃的0.78nm增大到900℃的1.08nm,可见WTAC-K的微孔在900℃下逐渐扩大并向中孔结构过度。
WTAC-Zn在 400℃下的总比表面积最大,达1596m2/g,继续增大活化温度BET逐渐减小。通过观察孔容量可发现,WTAC-Zn-500-1(1)的微孔容量最小而中孔容量最大,可见随温度的升高,WTAC-Zn的微孔逐渐扩大进化为中孔,而继续升高温度材料的孔结构因烧失而坍塌并重排,得到新的微孔结构,当温度达 700℃时,几乎不存在中孔结构。此外,在600℃之前WTAC-Zn的平均微孔径随温度的升高而增大,达600℃之后则随之缩小,由此可判断 600℃是 WTAC-Zn结构坍塌的临界温度。
2.4 活化时间对废茶活性炭孔结构的控制
固定废茶与K2CO3在浸渍比为1∶1,在700℃下活化0.5~3.0h,固定废茶与ZnCl2在浸渍比为1∶1,在400℃下活化0.5~2.0h,研究了活化时间对活性炭产率及结构的影响。如图5为活性炭的产率随活化时间变化的趋势线,由两种材料的趋势线可以看出,随着时间的延长材料组分进一步分解、去除,活性炭的产率逐渐降低,但是二者产率降低的趋势并不明显。
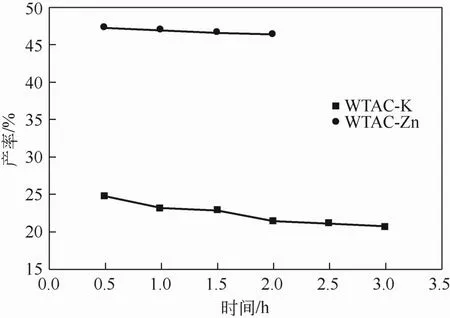
图5 WTAC产率随活化时间变化趋势图
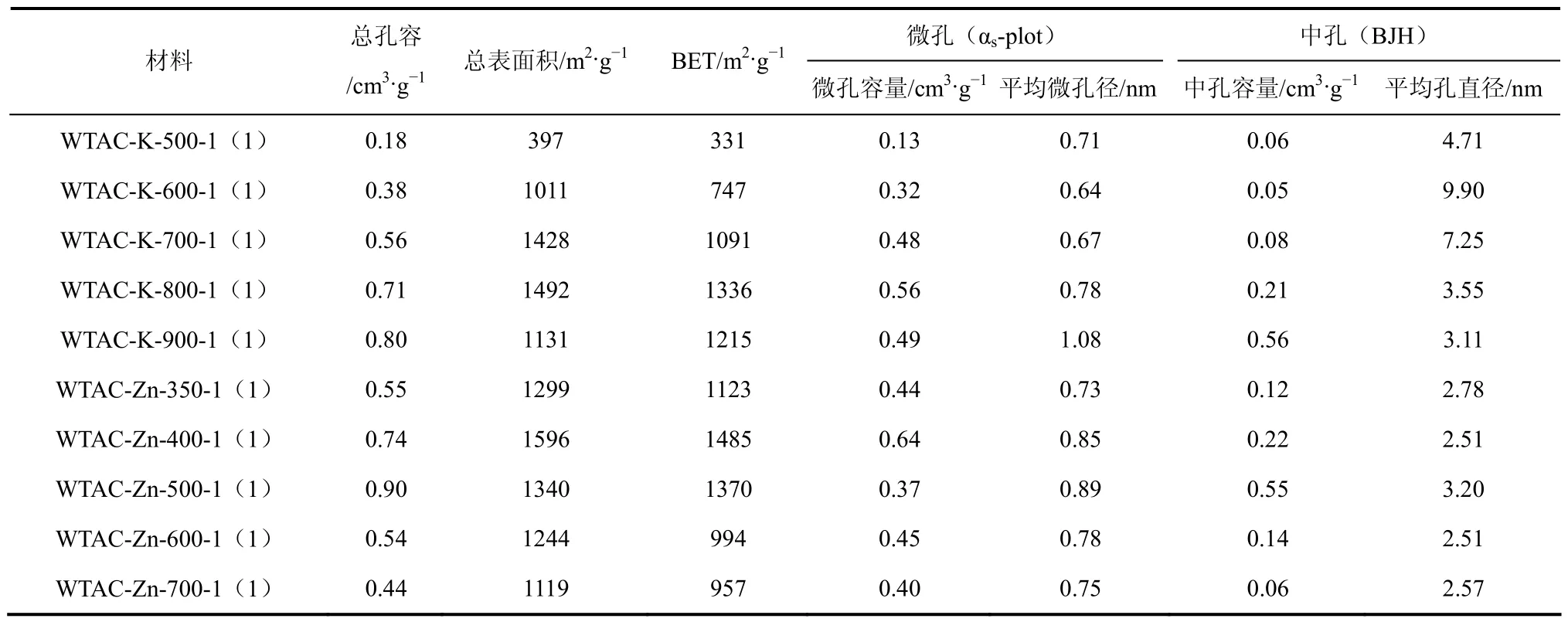
表3 活化温度对孔结构的影响
图6为不同活化时间下WTAC的氮气吸附等温线,WTAC-K的等温线均符合I-B型吸附等温线类型,说明WTAC-K材料以微孔结构为主导,并存在一定的大孔结构,0.5~1.5h的活化时间对废茶活性炭的氮气吸附等温线影响不大,三者的吸附等温线几乎完全重合,当活化时间提高到2h后,其氮气饱和吸附量明显提高。当活化时间达到 1.5h后WTAC-Zn的氮气吸附等温线开始由Ⅰ型向Ⅳ型转变并伴随迟滞环的出现,可见存在一定的中孔结构,继续延长活化时间,其等温线又转变为Ⅰ型,迟滞环消失。
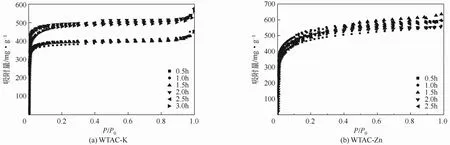
图6 不同活化时间下废茶活性炭的氮气吸附等温线
不同活化时间下制备 WTAC的孔径结构参数见表4,以K2CO3为活化剂时,0.5~1.5h的活化时间对WTAC-K的比表面积影响不大,当活化时间达到2h后,其BET比表面及微孔容量显著提高,继续延长活化时间至2.5h,中孔容量提高。此外,在0.5~2.5h,平均微孔径几乎不变,均为 0.67nm左右,当活化时间延长至3h,微孔径开始增大,并向中孔结构过度。
ZnCl2为活化剂时,WTAC-Zn-400-1(1)的BET比表面积与微孔容量最大,当活化时间达1.5h时微孔容量降低,中孔容量提高,继续延长活化时间至2h后,中孔容量降低,微孔容量升高,微孔径有所收缩。说明随着活化时间的延长,WTAC-Zn的微孔径逐渐扩大并过渡到中孔,继续延长活化时间使炭结构重排,孔径收缩得到新的微孔结构。
2.5 浸渍比对废茶活性炭孔结构的控制
固定活化活化温度及活化时间,研究废茶与活化剂浸渍比例对 WTAC产率及结构的影响。其中K2CO3与废茶在0.5、1、2等3种比例下700℃活化1h,ZnCl2与废茶在0.5、0.8、1、1.2等4种比例下400℃活化1h。图7为WTAC的产率随浸渍比例变化的趋势线,产率随浸渍比例的提高逐渐下降,主要是因为在活化过程中,增大活化剂浓度,可以促使活化剂对材料表面的刻蚀作用,从而使得废茶活性炭产率降低。
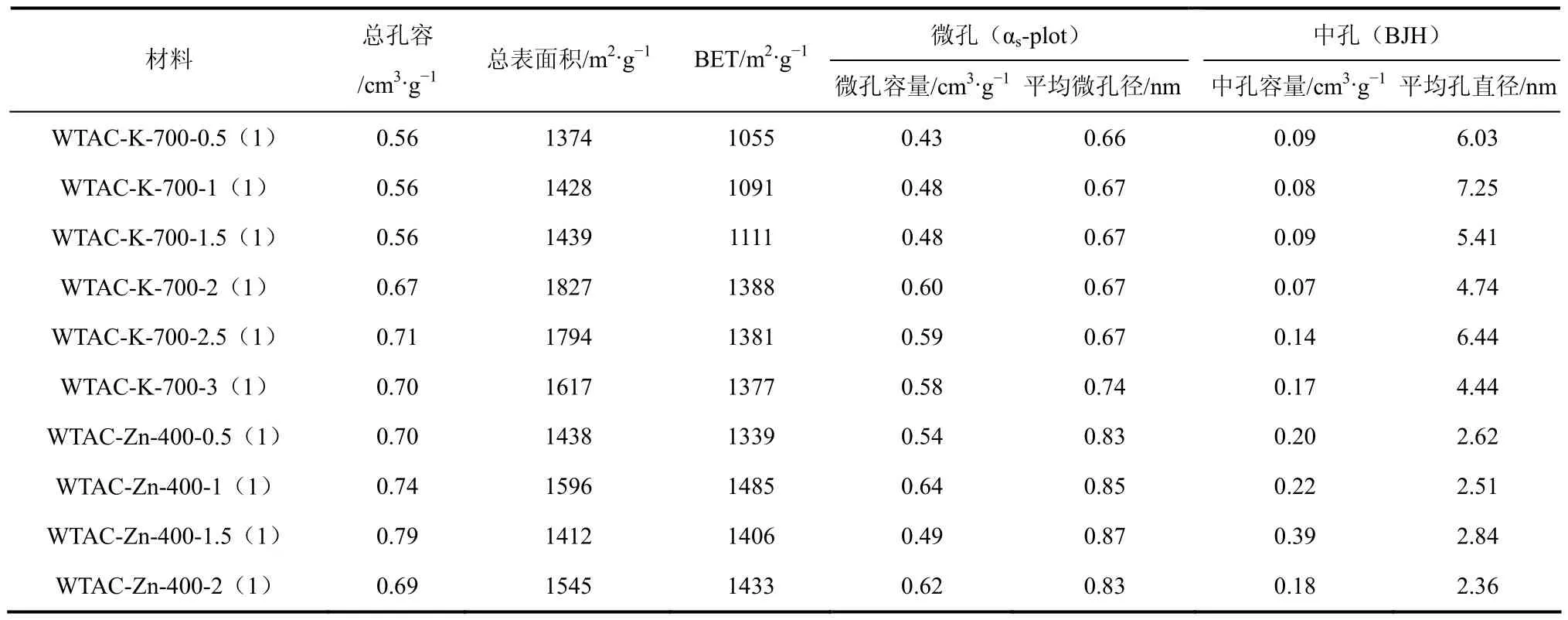
表4 活化时间对孔结构的影响
图8为不同浸渍比下废茶活性炭的氮气吸附等温线,不同浸渍比下的WTAC-K的等温线类型均符合Ⅰ-B型。在浸渍比为0.5~1.0时WTAC-Zn的吸附等温线类型符合Ⅰ-A型,以微孔结构为主,当浸渍比为1.2时,等温线类型发生了变化,并出现迟滞环,说明已有中孔结构形成。
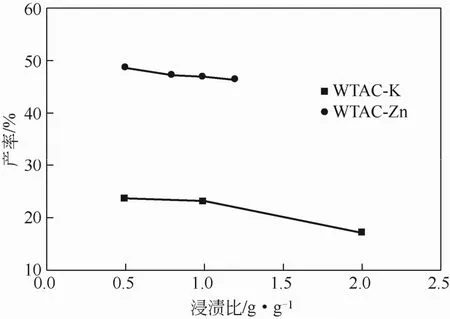
图7 WTAC产率随浸渍比例的变化趋势图
表5为不同浸渍比下制备的WTAC孔径结构参数,随着活化剂用量的提高WTAC-K的BET比表面积与微孔容量呈先增大后减小的趋势,其中浸渍比为1∶1时BET与微孔容量最大,可见当活化剂用量较少时,活化反应不够充分,形成的孔隙较少;当浸渍比为2∶1时,微孔容量及平均微孔径减小,这可能是由于较大的活化剂用量对炭材料的烧蚀作用较大,碳骨架的C被消耗,使其结构重排,因而得到较小的微孔结构。
WTAC-Zn的BET比表面积随浸渍比例的提高先增大后减小,其中1∶1的条件下WTAC-Zn的BET与微孔容量最大。随着浸渍比例的提高WTAC-Zn的微孔容量先增大后减小,而中孔容量逐渐升高,这主要是因为活化剂用量的增加使活化作用提高,因而孔结构越来越发达,当浸渍比超过1∶1时,所形成的微孔结构逐渐向中孔结构进化。由WTAC-Zn的平均微孔径也可以看出其微孔结构随活化剂用量的增加而增大。
3 结 论
(1)相比水蒸气物理活化,K2CO3、ZnCl2化学活化法更有利于废茶活性炭多孔性结构的形成;K2CO3、ZnCl2活化制备的废茶活性炭比表面积较大,最高能达1596m2/g,所制备的废茶活性炭均为微孔活性炭。
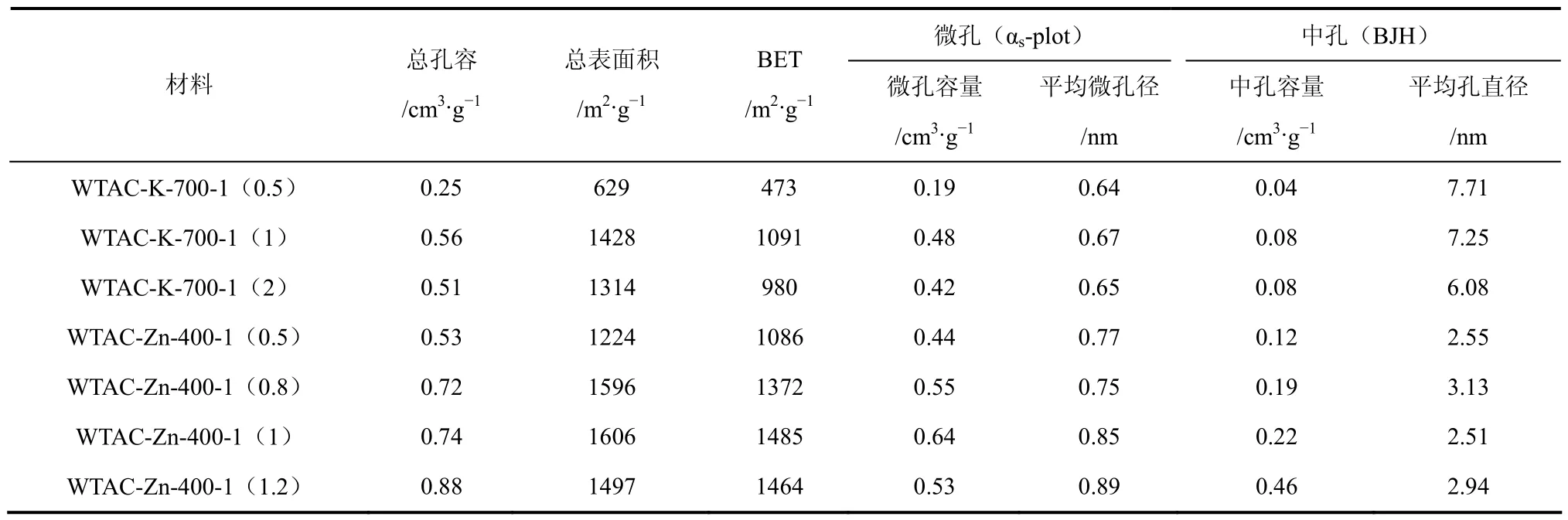
表5 浸渍比例对孔结构的影响
(2)较高的温度有利于WTAC-K孔结构的发展,当活化温度达到 800℃时,微孔容量最大,并开始产生中孔结构;WTAC-Zn在较低的活化温度下就已产生一定的微孔及中孔结构,当活化温度为400℃时,BET比表面积最大。
(3)活化时间对WTAC-K微孔结构的改变不大,主要是增加其比表面结构。随着活化时间的延长,WTAC-Zn产生新的微孔结构,当时间延长到1.5h,其微孔结构逐渐扩展为中孔结构,继续延长活化时间,由于过度烧失,孔结构得到重排得到更多的微孔结构。
(4)废茶活性炭的BET比表面积与微孔容量随浸渍比例的提高呈先增大后减小的趋势,其中两种活化剂均在浸渍比为 1∶1下所形成的微孔容量最大。
[1] 龚盛昭,叶孝兆,骆雪萍.利用废茶制备护肤霜的研究[J].广西化工,2002,31(1):12-13,26.
[2] Dem ir,I.An investigation on the production of construction brick w ith processed waste tea[J].Building and Environment,2006,41(9):1274-1278.
[3] Takahashi T, Aso Y, Kasai W,et al.Synergetic deodorant effect and antibacterial activity of composite paper containing waste tea leaves[J].Journal of Wood Science,2011,57(4):308-316.
[4] Panneerselvam P,Morad N,Tan K A.Magnetic nanoparticle(Fe3O4)impregnated onto tea waste for the removal of nickel(Ⅱ)from aqueous solution[J].Journal of Hazardous Materials,2011,186(1):160-168.
[5] 董宇,申哲民,王茜,等.生物质活性炭制备的比较研究[J].安徽农业科学,2011,39(6):3444-3448.
[6] Dias J M,Alvim-Ferraz M C M,Almeida M F,et al.Waste materials for activated carbon preparation and its use in aqueous-phase treatment:A review[J].Journal of Environmental Management,2007,85(4):833-846.
[7] Ioannidou O,Zabaniotou A.Agricultural residues as precursors for activated carbon production- a review[J].Renewable and Sustainable Energy Reviews,2007,11(9):1966-2005.
[8] Yagmur E,Ozmak M,Aktas Z.A novel method for production of activated carbon from waste tea by chemical activation with m icrowave energy[J].Fuel,2008,87:3278-3285.
[9] 刘靖,于家佳,邢殿楼,等.利用废茶制备活性炭及其吸附性能的研究[J].环境科学导刊,2011,30(1):1-4.
[10] Auta M,Hameed B H.Optim ized waste tea activated carbon for adsorption of methylene blue and acid blue 29 dyes using response surface methodology[J].Chemical Engineering Journal,2011,175:233-243.
[11] Gurten I I,Ozmak M,Yagmur E,et al.Preparation and characterization of activated carbon from waste tea using K2CO3[J].Biomass and Bioenergy,2012,37:73-81.
[12] Gundogdu A,Duran C,Senturk H B,et al.Adsorption of phenol from aqueous solution on a low-cost activated carbon produced from tea industry waste:Equilibrium,kinetic,and thermodynam ic study[J].Journal of Chemical & Engineering Data,2012,57(10):2733-2743.
[13] Borah,L,Senapati K K,Borgohain C,et al.Preparation of orderedporous carbon from tea by chemical activation and its use in Cr(Ⅵ)adsorption[J].Journal of Porous Materials,2011,19(5):767-774.
[14] Peng C,Yan X B,Wang R T,et al.Prom ising activated carbons derived from waste tea-leaves and their application in high performance supercapacitors electrodes[J].Electrochimica Acta,2013,87:401-408.
[15] Zhu Y,Gao J,Li Y,et al.Preparation of activated carbons for SO2adsorption by CO2and steam activation[J].Journal of the Taiwan Institute of Chemical Engineers,2012,43(1):112-119.
[16] Uçar S,Erdem M,Tay T,et al.Preparation and characterization of activated carbon produced from pomegranate seeds by ZnCl2activation[J].Applied Surface Science,2009,255(21):8890-8896.
[17] 赵朔,裴勇.氯化锌活化法制备笋壳基活性炭的工艺研究[J].材料导报,2012,26(4):87-90.
[18] Adinata D,Wan Daud W M A,Aroua M K.Preparation and characterization of activated carbon from palm shell by chem ical activation with K2CO3[J].Bioresource Technology,2007,98(1):145-149.
[19] Adinata D,Wan Daud W M,Aroua M K.Preparation and characterization of activated carbon from palm shell by chem ical activation w ith K2CO3[J].Bioresource Technology,2007,98(1):145-149.
[20] Dolas H,Sahin O,Saka C,et al.A new method on producing high surface area activated carbon:The effect of salt on the surface area and the pore size distribution of activated carbon prepared from pistachio shell[J].Chemical Engineering Journal,2011,166(1):191-197.
[21] Song L,Jun-ichi M,Hirofum i K,et al.Enhancement of the methylene blue adsorption rate for ultramicroporous carbon f ber by addition of mesopores[J].Carbon,2006,44(10):1884-1890.
[22] 罗来盛,孙利红,余阳,等.微波活化法制备加拿大一枝黄花活性炭及其性能表征[J].环境工程学报,2011,5(5):1161-1165.
[23] 田莹莹,刘恩辉,沈海杰,等.茶籽壳质活性炭的制备及其化学性能[J].功能材料,2012,43(6):752-755.
[24] 近藤精一,石川达雄,安部郁夫.吸附科学[M].李国希译.第2版.北京:化学工业出版社,2005.
[25] 刘小军,五永邦,乔文明,等.氯化锌活化制备沥青基球形活性炭[J].华东理工大学学报:自然科学版,2011,36(4):523-528.
[26] Olivares-Marín M,Fernándz-González C,Macías- García A,et al.Preparation of activated carbon from cherry stones by chem ical activation w ith ZnCl2[J].Applied Surface Science,2006,252(17):5967-5971.
[27] 张会平,叶李艺,杨立春.氯化锌活化法制备木质活性炭研究[J].材料科学与工艺,2006,14(1):42-45.
[28] Saka C.BET,TG-DTG,FT-IR,SEM,Iodine number analysis and preparation of activated carbon from acorn shell by chem ical activation w ith ZnCl2[J].Journal of Analytical and Applied Pyrolysis,2012,95:21-24.
[29] Özdem ir M,Bolgaz T,Saka C,et al.Preparation and characterization of activated carbon from cotton stalks in a two-stage process[J].Journal of Analytical and Applied Pyrolysis,2011,92(1):171-175.
[30] 周保华,高勤,郭斌,等.碳酸钾化学活化法制备土霉素菌渣活性炭研究[J].南京理工大学学报,2012,36(6):1070-1074.
[31] Horikawa T,Kitakaze Y,Sekida T,et al.,Characteristics and humidity control capacity of activated carbon from bamboo[J].Bioresource Technology,2010,101(11):3964-3969.
[32] 马承愚,熊慧珍,宋新山.ZnCl2活化茄子秸秆制备活性炭及表征[J].功能材料,2012,43(3):342-345.
[33] 王晓瑞,钱仁渊,金鸣林,等.活性炭制备工艺条件对其比表面积的影响[J].炭素技术,2004,23(6):1-4.
