1,3,4-噁二唑-2-硫酮衍生物的合成及工艺
2014-02-26巨修练
巨修练,胡 成
[1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程教育部重点实验室(武汉工程大学),湖北 武汉 430074]
0 引 言
杂环化合物自发现以来已有百年的历史,近年来随着结构分析与鉴定技术的进步,越来越多的杂环化合物被发现,杂环化合物的应用范围也在不断扩展,涉及到医药,农药,食品香料,化学染料等众多领域[1-2]. 噁二唑类衍生物作为杂环化合物中的一类,具有酶活性抑制[3],消炎[4],抗菌[5],抗癌等广泛的生物活性,并且具有高效低毒等优点;另外,由噁二唑化合物形成的聚合物具有特殊的稳定性,有些还具有良好的成膜性和电子传输性能,在材料领域也具有广泛的应用前景[6]. 因此,近年来噁二唑化合物的研究受到研究者的关注.孙娜波合成了一系列新型含吡唑环的1,3,4-噁二唑类衍生物并对其进行了活体杀菌活性测试,研究结果表明部分化合物对黄瓜褐斑病,黄瓜霜霉病和黄瓜菌核病均具有较好的防治效果,个别化合物在500 mg/L的剂量下对黄瓜褐斑病的防效高达90%,优于井冈霉素,故能作为先导化合物开发新农药[7]. Ghani等人研究了噻二唑,噁二唑和三唑杂环衍生物对酪氨酸酶的抑制效果,研究结果表明连接一定的疏水基团对这三类杂环的抑制活性有一定的提高,活性动力学研究表明噁二唑杂环5-C位上的取代基对分子与酪氨酸酶结合的紧密程度起着主要作用,5-C位上长链的存在对该类抑制剂活性的提高有重要作用,它能更好促进抑制剂与酶的结合. 其中5-对羟基苯-1,3,4-噁二唑-2-硫酮对酪氨酸酶的抑制常数Ki值可达1.77 μmol/L[3].Lam等[8]合成了一系列1,3,4-噁二唑-2-硫酮衍生物,并均表现出对酪氨酸酶的高效抑制作用,其IC50在0.87~1.49 μmol/L,均高于对照曲酸. 同时其结构与活性分析也证明了一定的疏水基团对该类化合物的活性有显著的提高.本研究通过图1所示合成路线合成了7个3位取代苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮化合物,期望得到更好的酪氨酸酶抑制剂,进而应用于医药,农药,化妆品等行业. 同时,所有目标化合物均通过氢核磁共振图谱和质谱表征确认.
1 实验部分
1.1 仪器与试剂
RY-1G型熔点仪;Varian Mercury-VX 300型核磁共振仪(CDCl3,DMSO为溶剂,TMS为内标);TRACEMS 2000型质谱仪.
所有试剂均为分析纯,购自于国药集团.反应过程采用的TLC薄层硅胶板和柱层析所用的硅胶,由青岛海洋硅胶干燥剂厂生产.CS2的纯化:采用质量分数5%的浓硫酸反复萃取至硫酸层为无色,再用蒸馏水洗涤CS2至中性,无水Na2SO4干燥,蒸馏收集48 ℃的馏分.其他试剂未经纯化直接使用.

图1 目标化合物的合成Fig.1 The synthetic routes of target compounds注:(a)NH2NH2·H2O,乙醇,回流;(b)CS2,KOH,乙醇,回流;(c)甲醛,胺,V(四氢呋喃)∶V(甲醇)=1∶2,室温搅拌.R为4-NO2(4a),3-NO2(4b),2-NO2(4c),2-OCH3(4d),2-Cl(4e),4-Cl(4f),2-COOCH3(4 g).
1.2 化合物的合成
1.2.1 苯甲酰肼的合成 向100 mL的三口烧瓶中依次加入苯甲酸甲酯(1)13.6 g (0.1 mol),质量分数80%水合肼18.75 g (0.3 mol),无水乙醇50 mL,升温至80 ℃回流,反应持续6 h,TLC监测无原料点,反应结束,冷却,旋蒸脱去溶剂,用少量乙醇重结晶得白色针状固体苯甲酰肼(2),干燥后称重为12.25 g,收率为90.1%.mp.112~114 ℃.
1.2.2 5-苯基-1,3,4-噁二唑-2-硫酮(3)的合成 向100 mL的三口烧瓶中依次加入苯甲酰肼(2)8.16 g (60 mmol)及KOH 4.032 g (72 mmol),CS26.84 g (90 mmol),无水乙醇60 mL,常温下搅拌0.5 h后,混合液呈白色乳状悬浊液,将混合液加热至80 ℃回流,反应持续8 h,TLC监测无原料点,反应结束,冷却,旋蒸脱去溶剂,将固体溶于蒸馏水,用3 mol/L的盐酸酸化至pH为4~5的得白色悬浊液,抽滤并将滤饼用乙醇重结晶得白色针状固体5-苯基-1,3,4-噁二唑-2-硫酮(3),称重为9.16 g,收率为85.8%,mp.218~220 ℃.1H NMR(400 MHz ,DMSO-d6) δ:7.56~7.65(m,3 H,ArH),7.86~7.88(d,2 H,ArH).
1.2.3 目标化合物3-对硝基苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4a)的合成 向50 mL的单口烧瓶中依次加入5-苯基-1,3,4-噁二唑-2-硫酮(3) 0.356 g (2 mmol),对硝基苯胺0.552 g(4 mmol),质量分数37%的甲醛溶液0.648 g(8 mmol),将其溶于THF和MeOH的混合液中[V(THF)=5 mL,V(MeOH)=10 mL],于常温下搅拌,0.5 h后开始出现黄色沉淀,反应进行4 h,TLC监测无原料点,反应结束,抽滤,滤饼用THF重结晶得暗黄色固体0.335 g,收率51.1%,mp.156~158 ℃.1H NMR(400 MHz,DMSO-d6) δ:5.51~5.53(d,2 H,—CH2—),6.95~6.97(d,2 H,ArH),7.47~7.55(m,3 H,ArH),7.76~7.78(d,2 H,ArH),7.98~8.00(d,2 H,ArH),8.32~8.36(t,1 H,—NH—); MS(ESI):327(M-1)-.
1.2.4 同样的方法合成化合物4b~4h 目标化合物3-间硝基苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4b):黄色固体0.512 g,收率78.0%,mp.181~182 ℃.1H NMR(400 MHz,DMSO-d6)δ:5.54~5.56(d,2 H,—CH2—),7.28~7.30(d,1H,ArH),7.37~7.41(t,1 H,ArH),7.47~7.49(d,1 H,ArH),7.54~7.61(m,3 H,ArH),7.80(s,1 H,ArH),7.82~7.84(d,2 H,ArH); MS(ESI):327(M-1)-.目标化合物3-邻硝基苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4c):黄色固体0.235 g,收率35.8%,mp.170~172 ℃.1H NMR(400 MHz,DMSO-d6)δ:5.73~5.75(d,2 H,—CH2—),6.84~6.88(t,1 H,ArH),7.39~7.42(d,1 H,ArH),7.51~7.61(m,4 H,ArH),7.83~7.85(d,1 H,ArH),8.08~8.10(d,1 H,ArH),8.79~8.82(t,1 H,—NH—); MS(ESI):327(M-1)-.目标化合物3-邻甲氧基苯胺亚甲基-5苯基-1,3,4-噁二唑-2-硫酮(4d):白色固体为0.286 g,收率45.7%,mp.134~136 ℃.1H NMR(400 MHz,CDCl3)δ:3.82(s,3 H,—OCH3),5.54~5.56(d,2 H,—CH2—),6.76(s,2 H,ArH),6.84~6.86(m,1 H,ArH),7.10~7.12(d,1 H,ArH),7.41~7.51(m,3 H,ArH),7.84~7.86(d,2 H,ArH); MS(ESI):312(M-1)-.目标化合物3-邻氯苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4e): 白色固体0.215 g,收率34.4%,mp.120~121 ℃.1H NMR(400 MHz,CDCl3)δ:5.56~5.58(d,2 H,—CH2—),5.66~5.70(t,1 H,—NH—),6.70~6.74(t,1 H,ArH),7.14~7.25(m,3 H,ArH),7.42~7.52(m,3 H,ArH),7.84~7.86(d,2 H,ArH); MS(ESI):317(M-1)-.目标化合物3-对氯苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4f):白色固体为0.159 g,收率25.4%,mp.216~218 ℃.1H NMR(400 MHz,CDCl3)δ:6.03(d,2 H,—CH2—),7.12~7.14(d,2 H,ArH),7.37~7.41(t,2 H,ArH),7.48~7.52(t,1 H,ArH),7.82~7.84(d,2 H,ArH),7.94~7.96(d,2 H,ArH); MS(ESI):317(M-1)-.目标化合物3-邻羧甲酯苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮(4g):白色固体0.458 g,收率67.2%,mp.158~160 ℃.1H NMR(400 MHz,CDCl3)δ:3.88(s,3 H,—COOCH3),5.60~5.62(d,2 H,—CH2—),6.73~6.77(t,1 H,ArH),7.27~7.29(d,1 H,ArH),7.41~7.47(m,3 H,ArH),7.50~7.54(t,1 H,ArH),7.88~7.90(d,2 H,ArH),7.92~7.94(d,1 H,ArH),8.83~8.87(t,1 H,—NH—);MS(ESI):340(M-1)-.
2 结果与讨论
2.1 苯甲酰肼(2)的合成工艺
2.1.1 原料配比对苯甲酰肼产率的影响 以无水乙醇为溶剂在回流条件下反应6 h,只改变质量分数80%水合肼和苯甲酸甲酯的配料摩尔比,粗产品用无水乙醇重结晶,结果如下:

表1 原料配比对产率的影响Table.1 Effect of raw material ratio on yield
结果表明质量分数80%水合肼与苯甲酸甲酯的配料摩尔比为3.0时,苯甲酰肼的产率最高,大于3.0时产率基本无变化.因此质量分数80%水合肼与苯甲酸甲酯的适原料摩尔比为3.0.
2.1.2 反应时间对苯甲酰肼产率的影响 以无水乙醇为溶剂,质量分数80%水合肼和苯甲酸甲酯的配料摩尔比为3∶1,改变反应时间,初产品用无水乙醇重结晶,结果如下:

表2 反应时间对产率的影响Table.2 Effect of reaction time on yield
结果表明当反应时间为6 h时,苯甲酰肼的产率高达90.1%,延长反应时间产率基本不变,基于节约能源方面的考虑,适宜的反应时间为6 h.
2.2 5-苯基-1,3,4-噁二唑-2-硫酮(3)的合成工艺
2.2.1 CS2用量对5-苯基-1,3,4-噁二唑-2-硫酮产率的影响 以无水乙醇为溶剂,苯甲酰肼和KOH摩尔比为1∶1.2,改变CS2的用量,回流反应8 h,按上述方法提纯,结果如下:

表3 CS2用量对产率的影响Table.3 Effect of dosage of CS2 on yield
结果表明二硫化碳与苯甲酰肼的配料摩尔比为1.5时,5-苯基-1,3,4-噁二唑-2-硫酮的产率高达85.8%,大于1.5时,产率基本不变.因此,二硫化碳与苯甲酰肼的适宜配料摩尔比为1.5.
2.2.2 反应时间对5-苯基-1,3,4-噁二唑-2-硫酮产率的影响 以无水乙醇为溶剂,苯甲酰肼,KOH,CS2的摩尔比为1∶1.2∶1.5,反应回流,改变反应时间,按上述方法提纯,结果为:
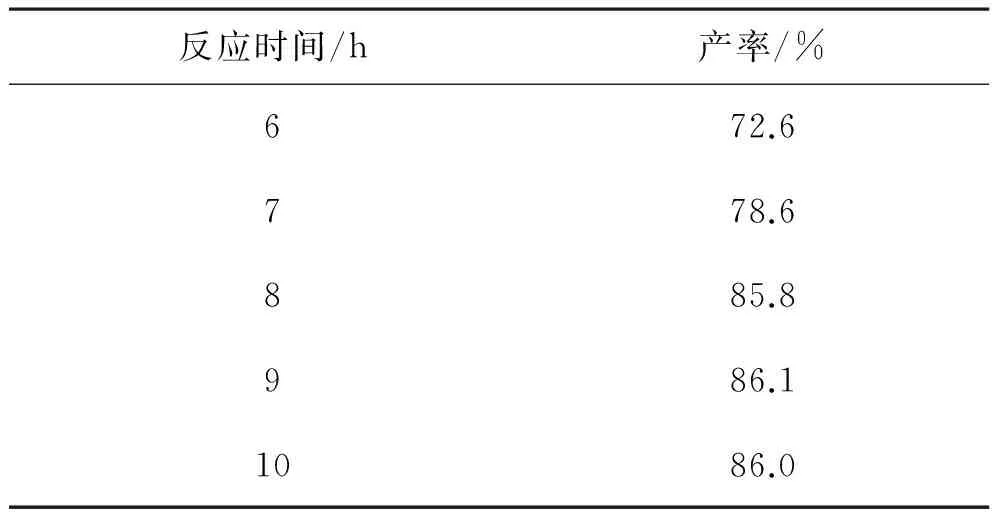
表4 反应时间对产率的影响Table.4 Effect of reaction time on yield
结果表明当反应时间为8 h时,5-苯基-1,3,4-噁二唑-2-硫酮的产率高达85.8%,延长反应时间产率基本不变,基于节约能源方面的考虑,适宜的反应时间为8 h.
2.3 3-取代苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮化合物的合成工艺
2.3.1 溶剂对3-对硝基苯胺亚甲基-5-苯基-1,3,4-噁二唑-2-硫酮产率的影响 按上述反应条件反应,改变所用溶剂(体积均为15 mL),粗产品重结晶,结果为:

表5 溶剂对产率的影响Table 5 Effect of solvent on yield
结果表明以V(甲醇)∶V(四氢呋喃)=2∶1的混合液为溶剂时,产率均高于单纯以甲醇或四氢呋喃为溶剂时的产率.以甲醇为溶剂时,反应过程中迅速得到絮状沉淀,使得5-苯基-1,3,4-噁二唑-2-硫酮(3)与目标产物共析导致大量原料包裹在絮状沉淀中,同时还影响反应液的搅拌,这些因素不仅会影响反应的产率,同时影响目标产物的分离;而以四氢呋喃为溶剂时,原料与目标产物均有较好的溶解性,导致析出的产物较少而影响产率.因此V(甲醇)∶V(四氢呋喃)=2∶1的混合液为较适宜的溶剂.
另外,本实验在以5-苯基-1,3,4-噁二唑-2-硫酮(3)与甲醛和胺发生Mannich反应的过程中先尝试了与仲胺吗啡啉反应预期得到对应的Mannich碱,然而结果生成的产物却不是预期的化合物,而是5-苯基-1,3,4-噁二唑-2-硫酮(3)在3-N为发生羟甲基化,吗啡啉并没有接到5-苯基-1,3,4-噁二唑-2-硫酮(3)上.这可能是因为5-苯基-1,3,4-噁二唑-2-硫酮(3)的亲和性比吗啡啉强,而易于与甲醛先发生亲和反应,得到的羟甲基化产物与吗啡啉脱水得到Mannich碱的反应能垒又太高,常温下难以进行而无法得到预期产物.
3 结 语
本研究通过参考相关文献和资料,设计在5-苯基-1,3,4-噁二唑-2-硫酮的3-N位通过发生Mannich反应引入一定疏水基团,对其进行结构修饰,通过核磁共振氢谱和质谱结构表征,合成了4a~4g共7个目标化合物,大部分未见文献报道,同时对反应中的原料配料比,反应时间及溶剂选择等参数进行了工艺优化.后续将检测目标化合物对酪氨酸酶的抑制活性,期望得到高效的酪氨酸酶抑制剂,以用于医药,农药及化妆品等领域.
致 谢
衷心感谢深圳海王药业对该项目研究提供的资助!
[1] 陶绍木,张建华,彭昌亚.杂环化合物的应用和发展[J].中国食品添加剂,2003(3):31-34. TAO Shao-mu,ZHANG Jian-hua,PENG Chang-ya.The application and development of heterocyclic compounds[J].China Food Additives,2003(3):31-34.(in Chinese)
[2] 巨修练,杨诗宏.苯并噻唑类衍生物的合成[J].武汉工程大学学报,2013,35(4):11-13. JU Xiu-lian,YANG Si-hong.Synthesis of benzothiazole derivatives[J].Journal of Wuhan Institute of Technology,2013,35(4):11-13.(in Chinese)
[3] GHANI U,ULLAH N.New potent inhibitors of tyrosinase:novel clues to binding of 1,3,4-thiadiazole-2(3H)-thiones,1,3,4-oxadiazole-2(3H)-thiones,4-amino-1,2,4-triazole-5(4H)-thiones,and substituted hydrazides to the dicopper active site[J].Bioorganic & Medicinal Chemistry,2010,18(11):4042-4048.
[4] GILANI S J,KHAN S A,SIDDIQUI N.Synthesis and pharmacological evaluation of condensed heterocyclic 6-substituted 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazole and 1,3,4-oxadiazole derivatives of isoniazid[J].Bioorganic & Medicinal Chemistry Letters,2010,20 (16):4762-4765.
[5] NAVEENA C S,BOJA P,KUMARI N S.Synthesis,characterization and antimicrobial activity of some disubstituted 1,3,4-oxadiazoles carrying 2-(aryloxymethyl)phenyl moiety[J].European Journal of Medicinal Chemistry,2010,45(11):4708-4719.
[6] 冯国仁 ,陶兰,刘影英.1,3,4-噁二唑衍生物的合成研究进展[J].杭州师范学院学报:自然科学版,2006,5(6):469-475. FENG Guo-ren, TAO Lan,LIU Ying-ying.Progress in the synthesis of 1,3,4-oxadiazoles[J].Journal of Hangzhou Teachers College:Nature Science Edition,2006,5(6):469-475.(in Chinese)
[7] 孙娜波,童建颖,武宏科.含吡唑环的1,3,4-噁二唑类衍生物的合成及杀菌活性研究[J].有机化学,2012,33(1):101-105. SUN Na-bo,TONG Jian-ying,WU Hong-ke.Synthesis and fungicidal activity of 1,3,4-oxadiazole derivatives containing pyrazole moiety[J].Chinese Journal of Organic Chemistry,2012,33(1):101-105.(in Chinese)
[8] LAM K W,SYAHIDA A, UI-HAQ Z,et al.Synthesis and biological activity of oxadiazole and triazolothiadiazole derivatives as tyrosinase inhibitors[J].Bioorganic & Medicinal Chemistry Letters,2010,20(12):3755-3759.
