六芳基联咪唑分子开关的研究进展
2014-02-23龚文亮熊祖劲朱明强
龚文亮,熊祖劲,朱明强
(武汉光电国家实验室,华中科技大学 光学与电子信息学院,湖北 武汉430074)
光致变色现象是指一个化合物(A)在受到一定波长的光的照射时,可以进行特定的化学反应,获得产物(B),由于结构的改变导致其吸收光谱发生明显的变化;同时,在一定的条件下(如热,光照等),产物(B)可以回到初始化合物(A)[1]。在英语中,光致变色的单词为“Photochromism”,对其定义 为 “light-induced reversible change of color(光诱导的可逆的颜色变化)”[2]。光致变色现象最早于1867年被Fritzsche报道[3]。他在研究并四苯(tetracene)时发现,并四苯溶液在光照下由橘红色变为无色,而在黑暗中则会自动由无色返回到橘红色。之后,各种光致变色材料,包括有机分子和无机材料,陆续被发现。在有机光致变色材料中,发展研究的比较成熟的主要有螺吡喃、螺噁嗪、苯并吡喃、俘精酸酐以及二芳基乙烯等。其中,作为新生代的光致变色材料六芳基联咪唑(Hexaarylbiimidazole,简称 HABI,注:在文章后面如不特别说明都用HABI代替Hexaarylbiimidazole或者六芳基并咪唑)于1960年被日本科学家Hayashi和 Maede在研究2,4,5-三苯基咪唑氧化的化学发光过程中被发现[4]。不同于一般的光致变色材料,HABI同时具备光致变色、压致变色以及热致变色性能。自从1960年HABI被发现以来,立即引起了科学家们广泛的研究兴趣。关于HABI的研究直至今日仍然处于初期阶段,而将HABI作为光致变色材料的研究也不是很多。因此,为了更加全面的了解HABI这种新型的光致变色材料的发展历程,本文将以时间前后为顺序,介绍在HABI发展过程中起到重要作用的文献。至于将HABI作为光致变色材料以外的研究(如聚合反应中的引发剂)以及关于HABI专利的内容将不在本文中涉及。
1 早期HABI的研究(1960-1970)
1960年(March 7th,文章的接收日期),Taro Hayashi和 Koko Maede在 The Journal of Chemical Physics期刊上发表了一篇标题为:“A New Phototropic Substance and Its ESR”的 研 究 论文[4]。他们利用K3Fe(CN)6和 KOH 的水溶液作为氧化剂,室温下氧化2,4,5-三苯基咪唑(TPI)得到了能够在紫外以及自然光照下变色的新型光致变色材料。通过对产物进行元素分析,他们提出这种光致变色材料是两分子的2,4,5-三苯基咪唑失去咪唑环N—H上的H,然后结合形成的二聚体(Dimer),而由光照后有色态的ESR研究得出其光致变色的机理。2,4,5-三苯基咪唑二聚体,即六芳基联咪唑(HABI),在紫外及近紫外光照下,六芳基联咪唑分子中连接两个咪唑环的氮-氮共价键被均裂打断,形成了2,4,5-三苯基咪唑自由基(triphenylimidazole radical,TPIR),其有色态的热褪色服从一级动力学机理。另外,他们还发现这种新型的光致变色材料还具备有热致变色性能。同年四月,他们在期刊Bulletin of the Chemical Society of Japan上报道了 HABI合成的详细步骤[5],并给出了光致变色生成的自由基的结构(如图1)。
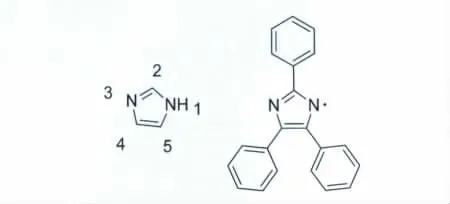
图1 Hayashi和Maede给出的2,4,5-三苯基咪唑自由基的化学结构式(附:咪唑化学位点的命名)Chemical structure of triphenylimidazole radical described by Hayashi and Maede(inset:nomenclature site in imidazole)
1961年Zimmermann在期刊Angewandte Chemie上独立报道了HABI的合成,同时提出了HABI的化学结构式以及光致变色机理(图2)[6]。

图2 Zimmermann给出的HABI的结构以及其光致变色的机理Chemical structure of HABI and its photochromism described by Zimmermann
1964年,Hisashi Ueda在期刊 The Journal of Physical Chemistry上发表了研究论文,详细的研究了2,4,5-三苯基 咪唑自 由基的 ESR 性 质[7]。同年,Hayashi和 Maeda在期刊 Bulletin of the Chemical Society of Japan上发表了研究论 文[8],他们利用2,4,5-三苯基咪唑自由基ESR信号,得出其有色态的热褪色动力学为二级动力学,修改了之前作出其褪色为一级动力学的判断。之后,在1966年,M.A.J.Wilks和 M.R.Willis在期刊Nature上发表了研究论文,总结了之前的工作,系统的推导出2,4,5-三苯基咪唑自由基热褪色为3/2级动力学[9]。
1966年,D.M.White和J.Sonnenberg在期刊Journal of the American Chemistry Society上发表了研究论文,系统讨论了HABI分子与光致变色直接相关的可能结构[10]。他们通过化学合成手段的控制,实现了两种二聚体(即文中的化合物2与化合物3)的相互转化,同时文中指出,HABI作为光致变色材料的结构是化合物3(即一个咪唑环的1号位氮上的碳与另一个咪唑环的2’号位碳原子相连的结构)(化合物2和3的结构如图3所示)。

图3 D.M.White和J.Sonnenberg在文章中给出的化合物1~4的结构式Chemical structure of compounds 1—4 described by D.M.White and J.Sonnenberg
1971年,Robert.L.Cohen在期刊Journal of Organic Chemistry上发表了研究论文[11]。文中提到通过对2-(o)Br-HABI的晶体衍射结果进行分析,证实了D.M.White和J.Sonnenberg关于HABI结构的结论。自此关于HABI作为光致变色材料以1-2’的结构存在被广泛的接受。(注:原文中关于晶体衍射的结果的引用文献为G.Teufer,private communication in advance of publication)。
1972年,Hideo Tanino等在期刊 Bulletin of the Chemical Society of Japan 上 发 表 了 研 究 论文[12],利用刚刚新兴的核磁技术比较全面的讨论研究了HABI各种可能结构的核磁图谱。
综合上述研究工作,我们大致可以归纳出HABI作为新型光致变色材料的如下特征:
1.1 光致变色的反应式
从早期的研究工作来看,关于HABI的具体结构一直有不同的争论。最初Hayashi和Maeda还有Zimmermann都提出两个咪唑环之间通过N—N相连的结构。后来随着D.M.White和J.Sonnenberg的研究工作以及晶体衍射技术的分析,得出HABI光致变色的结构应该是1(N)-2’(C)相连的方式,自此关于HABI的结构被广泛的接受。HABI的合成方法以及其光致变色的反应式见图4。关于不同的取代基团对于HABI有色态的吸收波长、摩尔吸光系数以及逆反应的热回复常数的影响,我们主要摘引Rolf Dessauer出版的著作中关于HABI的介绍(书名为Photochemistry,History and Commercial Applications of Hexaarylbiimidazole)[13]。
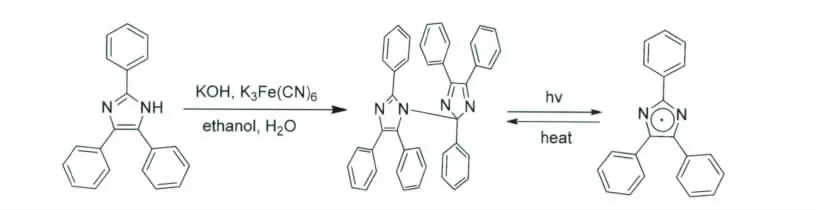
图4 HABI的制备方法以及其光致变色的化学反应式Synthesis and photochromism of HABI
1.2 不同的取代基团对于三苯基咪唑自由基(TPIR)最大吸收波长(颜色)的影响
如图5,从合成上看,对于HABI的改性可粗略分为两部分:第一部分是2号位上的苯环,与之直接对应的是合成2,4,5-三苯基咪唑的芳香醛(图中红色部分);第二部分是4,5号位上的两个苯环,与之直接对应的是原料中的苯偶酰(图中绿色部分)。大量的研究结果表明,在HABI的4,5号位上进行改性对于HABI有色态的吸收波长、逆反应回复速率等影响较小;而对2号位的苯环的改性则有比较大的影响。因此,我们重点讨论通过对2号位苯环引入不同的基团对于三苯基咪唑自由基(TPIR)的光谱以及动力学性质的影响。书中讨论的各种基团详见图5。

图6 不同的取代基对于三苯基自由基(TPIR)的最大吸收波长的影响Effect of different substituted groups on maxima absorption wavelengths of TPIRs
图6 是横轴为不同取代基的三苯基自由基(TPIR)的最大吸收波长。当无取代基团,即最简单的TPIR时,其最大吸收波长为550nm。当R=(4)-NO2时,最大吸收波长蓝移到530nm,而当R=(4)-OCH3时,最大吸收波长则红移到610 nm。当取代基团相同,氯原子处于对位时,λmax=570nm;氯原子处于间位时,λmax=550nm;氯原子处于邻位时,λmax=540nm。当 R=(4)-OCH3时,λmax=610nm,而当R=(2,4)di-OCH3,λmax=620 nm。由以上结果可得出如下结论:当R为拉电子基团时,λmax向短波方向移动,而当R为推电子基团时则向长波方向移动。取代基团相同位置不同时,处于对位时由于对自由基的共轭作用更明显,其最大吸收波长相对于邻位更加红移;间位由于没有明显的共轭作用对最大吸收波长影响不大,邻位则由于存在一定的扭曲结构缩短了自由基的共轭而使最大吸收波长略有蓝移。当推电子基团越多,最大吸收波长的红移越明显。
1.3 不同的取代基团对于二聚体复合常数的影响
不同的取代基团对于二聚体复合常数的影响总结于图7中。在溶剂苯中,R=H时,其二聚体复合 常 数 (Dimerization Constant,k)为 7.7×10-3。当取代基团处于苯环的对位上时,其二聚体复合常数都相对于无取代基团的时候略小或者相当(R=CN,k=10×10-3),而与取代基团的推拉电子属性无关。而当取代基团处于间位时,二聚体复合常数相比无取代基团时有小幅增大(3-NO2,k=12.8×10-3)。当取代基团处于邻位时,二聚体复合常数则有明显的提升。其中o-Cl-HABI的二聚体复合常数k达到122×10-3,是无取代基团时候的15.8倍。因此在2号位苯环的邻位上引入适当的基团能够使得HABI作为分子开关的热回复速率得到大幅度的提升。

图7 不同的取代基对于三苯基自由基二聚体复合常数(k)的影响Effect of different substituted groups on dimerization constant of TPIRs
1.4 不同的取代基团对于2,4,5-三苯基咪唑自由基摩尔吸光系数的影响
不同的取代基团对于2,4,5-三苯基咪唑自由基摩尔吸光系数的影响总结于图8中。总体来看,当取代基团为拉电子基团时,摩尔吸光系数会变小;而当取代基团为推电子基团时,其摩尔吸光系数会变大。
2 Jiro Abe课题组的研究工作(1999-2013)
自从HABI于1960年初被日本科学家Hayashi和Maede发现以来,将其作为光致变色材料的研究立即引起了科学家的关注。HABI同时具有光致变色,热致变色以及压致变色性能,这是一般的分子开关很难同时具备的。但是由于其光致变色之后的逆反应的恢复速率较慢(其热褪色的半衰期τ1/2长达数分钟,乃至数十分钟),同时光致变色后的产物为活泼的自由基,使得其抗疲劳性较差,因此,很少有人将HABI作为分子开关来研究,而主要研究也集中于作为聚合物的引发剂以及照片显影(Photoimaging)。

图8 不同的取代基对于三苯基自由基摩尔吸光系数的影响Effect of different substituted groups on molar absorption coefficients of TPIRs
HABI在光致变色之后的光消色反应之所以慢,主要由于光致变色之后产生的两个2,4,5-三苯基咪唑自由基(TPIR)相互分离。而光消色的过程相当于两分子的自由基相互结合的反应。由于溶剂的对流以及阻隔作用,其热恢复过程很慢也就不足为奇了。在有机化学中,自由基是活泼性极高的物种,这样HABI在光致变色之后如果不能很快以两分子复合消色而回到初态,那么它很容易和溶剂中的O2或者与可能存在的活泼氢作用而失去光致变色性能。综合这些原因,传统的HABI分子作为分子开关的抗疲劳性一般比较差。
我们知道对于任何一个分子开关而言,多数情况下,我们总希望它在受到外界刺激(如光、电、热、声、压力等)的时候能够迅速的回到初始态。一方面这是实际应用的需要,另一方面这对于分子开关的抗疲劳性也很重要。因为多数的光致变色材料的变色是通过在外界刺激下破坏分子开关中原有的化学键(如螺吡喃,螺噁嗪等)或者改变构象(如偶氮苯),而去除外界刺激之后,化学键又重新复合的过程来实现的。而在原有化学键被破坏的同时,生成的中间体的化学反应活泼性通常都比较高,很容易在这个过程中与介质中的其他分子(如空气中的O2)相互作用而“变质”,失去了分子开关的性能。因此提高分子开关材料在受到外界刺激变色后回到初始态的速率对于一个光致变色材料至关重要。近十几年,Jiro Abe课题组的一系列工作使HABI作为光致变色材料应用由理论变为了可能,因此本部分主要以Jiro Abe课题组近十几年的研究工作为主线,对HABI这种新型的光致变色材料进行介绍。
Jiro Abe课题组在研究tF-BDPI-2Y(其分子设计的结构见图9Chart 2)时发现,该分子为闭合的结构,其光致变色过程在理论上存在两个断键的过程。如果只让其中一端无法在光照过程中断键,那么就能达到限制两分子TPIR的自由活动,进而提升其逆反应速率的目的。据此他们提出将两个三苯基咪唑自由基“绑”在一起的分子结构设想。2007年,他们通过在萘的1,8-位上同时引入两个三苯基咪唑,合成了分子1,8-TPID-naphthalene[14](如图9)。在光照之后,两个咪唑环之间的C—N键断裂生成两个TPIR,由于它们被固定在萘环上,无法自由扩散,避免了溶剂分子的阻隔作用,相互之间的碰撞的几率极大的提高,使得逆反应的速率得到了极大的提升(τ1/2=2.04s,295 K),且该逆反应服从化学反应的一级反应动力学过程。他们通过萘桥联限制产生的自由基的“行动”,避免了两分子的TPIR在溶剂中扩散后“相遇”难的问题,极大地加快了HABI逆反应,即两个自由基形成HABI的热回复反应的速率,使得HABI有色态的热褪色速率由数分钟降低至2s,实现了材料性能上质的飞越。

图9 传统的HABI分子,tF-BDPI-2Y的分步光致变色过程以及桥联绑定 HABIPhotochromism of conventional HABIs,bi-HABI(tF-BDPI-2Y)and diffusion-inhibited HABI
之后,在1,8-TPID-naphthalene分子设计的基础上,如果一个分子中的咪唑环不是两个而是四个那么对其逆反应有什么影响呢?2008年,在1,8-TPID-naphthalene 分 子 设 计 的 基 础 上 Jiro Abe课题组合成了分子1C[15]。分子1C中有两个咪唑环,可以看做一个分子中有“两组”HABI。由于光致变色过程存在两步,即1C需经过1B再到1A,因此从1A返回到1C需要经过一个中间过程,使得其光致变色的逆反应不是一级动力学过程。最终的结果是其热恢复相对于1,8-TPIDnaphthalene要慢一些。
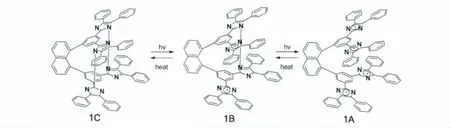
图10 1C的分子结构式以及光致变色的反应式Molecular structure and photochromism of 1C
因此,从实用的角度来看一个分子中设计多个(超过2个)HABI是不可取的。2008年,该课题组继续报道了改性的1,8-TPID-naphthalene,即1,8-NDPI-TPI-naphthalene[16]。当1,8-TPIDnaphthalene分子中与萘直接相连的一个苯环替代为萘之后,在295K的条件下,其热消色的半衰期(τ1/2)由2.04s缩短到0.26s,为原来的十分之一!实现了激光笔指在哪儿,哪儿变色,移动激光笔马上消色,而且肉眼无法区分的效果(其分子结构以及光致变色效果见图11)!虽然其中的原因至今仍不明确,但是从分子设计的角度来看,一个细小的改进就取得如此显著的效果,显示了HABI作为新型的光致变色材料具有极其广阔的研究空间以及应用前景。

图11 1,8-NDPI-TPI-naphthalene分子结构式、光致变色的反应式以及光致变色效果图Molecular structure and photochromism of 1,8-NDPI-TPI-naphthalene
以上基于萘环1,8位紧邻的结构特点来“捆绑”两个三苯基咪唑自由基的分子结构设计极大的改善了HABI作为分子开关的应用前景。然而,如果将HABI分子开关以视频帧的速率应用于即时成像中时,必须要求分子开关的消色速率在几十毫秒内完成,而最快的1,8-NDPITPI-naphthalene的消色速率为260ms,勉强能够满足要求。如何才能更进一步的提升HABI在变色后的消色速率呢?Jiro Abe课题组设计合成了以对环芳烃(Paracyclophane)取代萘的新的 分 子,pseudogem-bisDPI[2.2]PC[17]。在 298 K的温度下,其消色半衰期(τ1/2)为33ms,完全足够应用于即时成像(其分子结构以及光致变色反应式见图12)。同时在O2存在的条件下,其分子开关的变色与消色可重复数千次而没有任何的退化,显示出了良好的抗疲劳性以及极好的应用前景。

图12 pseudogem-bisDPI[2.2]PC分子结构式以及光致变色的反应式Molecular structure and photochromism of pseudogem-bisDPI[2.2]PC
在pseudogem-bisDPI[2.2]PC 分子的基础上,将其中一个咪唑环的4号位以及5号位的两个苯环闭合起来形成菲并咪唑的结构时得到的pseudogem-DPI-PI[2.2]PC 分子(其分子结构以及光致变色反应式见图13),其在室温下的有色态褪色半衰期(τ1/2)为35μs[18]。这几乎是pseudogem-bisDPI[2.2]PC分子有色态褪色半衰期的十分之一!他们认为当一个咪唑环上的两个苯环被闭合形成菲并咪唑之后,改变了热褪色过程中两个自由基的吉布斯自由能(),并由理论计算证明了这一推论(具体的理论参数以及能级图见图13和表1)。值得注意的是,在这种分子设计中,如果上下都是菲并咪唑则不具有光致变色性能。这与之前传统HABI分子,在咪唑环4,5号位上的两个苯环如果闭合后也将失去光致变色性能不谋而合[19],而其中的原因依然未知。
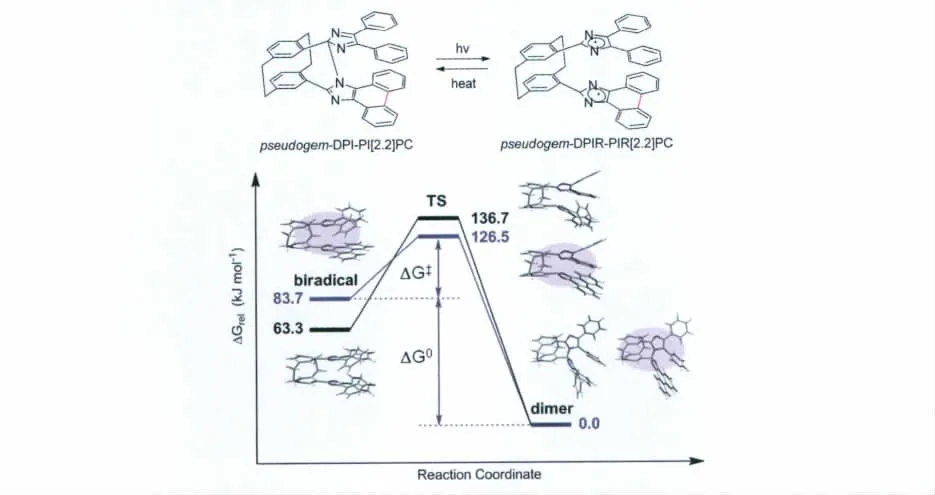
图13 pseudogem-DPI-PI[2.2]PC的光致变色化学反应式;pseudogem-bisDPI[2.2]PC和pseudogem-DPI-PI[2.2]PC由 DFT M05-2X/6-31G(d)计算获得的其热回复过程的能级图;蓝色部分代表简化的pseudogem-DPI-PI[2.2]PC结构Molecular structure and photochromism of pseudogem-DPI-PI[2.2]PC;energy levels of pseudogem-bisDPI[2.2]PC and[ ] / (); : [ ]
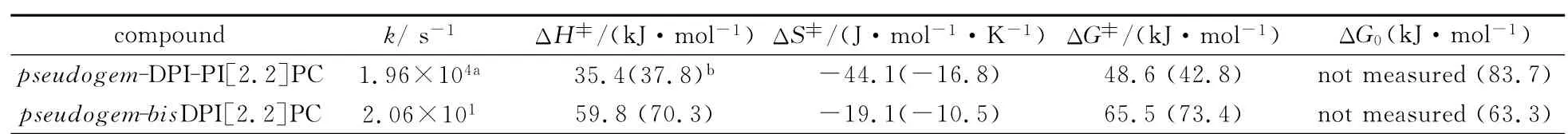
表1 298K 下,pseudogem-bisDPI[2.2]PC和pseudogem-DPI-PI[2.2]PC热回复反应的动力学和热力学参数Thermo-induced decoloration reaction kinetic and thermodynamic parameters of pseudogem-bisDPI[2.2]PC and pseudogem-DPI-PI[2.2]PC at 298K
由于Jiro Abe课题组的不断研究使得HABI作为分子开关热回复的半衰期由原来的数分钟降低至35μs!由此我们可以看到对HABI分子进行合理的设计可以使得其作为光致变色材料有质的飞越,显示出了巨大的潜力。在此我们继续介绍一些特殊的桥联绑定(Diffusion Inhibited)HABI分子。2012年,Jiro Abe课题组以1,1’-联二萘为桥联结构将两个三苯基咪唑绑定在其2,2’位上(见图14),得到了具有负光致变色性能的HABI[20]。由图14可以看到,氧化生成的2,2’直接相连的产物(无色态)由于化学键的扭曲太大,在热力学上极不稳定,很容易通过生成自由基然后形成以一个咪唑环的1号位N与另一个萘环的1号位直接相连的有色态形式,或者直接由热力学过程变为有色态形式。这一研究结果丰富了HABI作为光致变色材料的多样性。
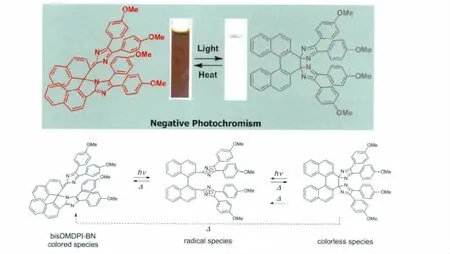
图14 bisDMDPI-BN分子的负光致变色化学反应式以及变色效果图Negative photochromism of bisDMDPI-BN
最近,即2013年,该课题组在原先1,8-TPIDnaphthalene分子设计的基础上,将两个三苯基咪唑由“头对头”的连接方式变为“头对尾”(如图15)[21]。通过改变两个三苯基咪唑的连接方式,他们发现两个咪唑环之间由原先的1-2’的N—C连接变为1-4’的N—C连接,这一研究结果说明HABI两个咪唑环之间的连接方式具有很大的可塑性。同时也发现反式的 1,8-bisTPI-naphthalene和1,8-bisTPI-naphthalene热褪色动力学性能没有太大的差异。
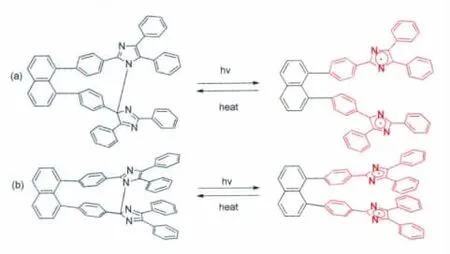
图15 (a)反式-1,8-bisTPI-naphthalene的分子结构以及其光致变色化学式;(b)1,8-bisTPI-naphthalene的分子结构以及其光致变色化学式Molecular structure and photochromism of trans-1,8-bisTPI-naphthalene(a)and 1,8-bisTPI-naphthalene(b)
我们通过上述研究工作继续深思:到底HABI分子中的两个咪唑环该如何连接呢?也许,我们能够从下面的一些研究工作中得到一些启示。2009年,Jiro Abe报道了将两种传统的HABI混合,在光照使其变色然后重新复合时发现原先的两种HABI分子在上述实验过程中变成了四种[22]。这说明传统的HABI分子在受到光照断键形成自由基的复合是随机组合的过程。2010年,他们又报道了新的HABI分子Pseudogem-DPIM-DPI[2.2]PC dimer(如 图16)[23]。我们从图16可以看到,虽然 Pseudogem-DPIM-DPI[2.2]PC dimer分子中有两个三苯基咪唑,可是其中的一个咪唑环上的NH被甲基取代为N-CH3,因此它无法被氧化继而在分子内形成化学键相互作用。因此Pseudogem-DPIM-DPI[2.2]PC只能通过两分子之间相互作用而不是之前的分子内的相互作用回到无色态。同时,研究发现两分子的Pseudogem-DPIM-DPI[2.2]PC之间并不是我们之前的一个咪唑环的1号N与另一个咪唑环2号C相连的形式(图16绿色分子结构式),而是对环芳烃苯环上的碳和咪唑环的N相连的形式(图16中蓝色分子结构式)。
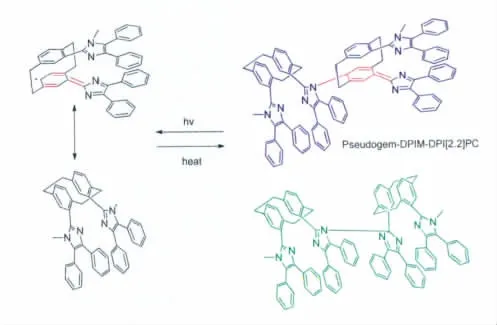
图16 Pseudogem-DPIM-DPI[2.2]PC dimer的光致变色反应式Molecular structure and photochromism of Pseudogem-DPIM-DPI[2.2]PC dimer
核磁共振技术对于现代有机合成,尤其是新合成产物的鉴别起着举足轻重的作用。Hideo Tanino等于1972年利用在当时刚刚兴起的核磁共振技术最早对HABI分子的核磁(氢谱)进行了分析。他们在文章中系统的探讨了HABI分子可能的六种结构。然而,一方面由于当时核磁技术还是处于刚刚起步,技术(比如13C-NMR以及二维-NMR等)不够成熟;另一方面,由于 HABI分子中的氢全部都是芳环氢,同时他们的化学环境相近,使得峰之间的相互重叠十分严重,而得到的氢谱看上去都是“一团乱麻”,很难仔细分辨。根据之前的研究工作我们知道HABI分子几种异构体由于能量差比较小,可以比较容易相互转换。因此,合成获得的HABI分子很难是单一结构的产物,更多的时候很有可能是几种结构共同存在的混合物。综合以上情况,HABI分子的核磁信息很多时候看起来很“杂乱”也就不足为奇了。由早期的研究工作我们知道,一般合成途径获得的HABI分子是以1-2’的连接方式为主,而在低温情况下(-70℃)1-2’连接的 HABI分子在照射形成自由基后其连接方式会变成以4-4’连接为主。这些研究结果的得出主要是基于其他技术(红外),而不是核磁技术。2013年,Jiro Abe课题组利用二维-NMR技术重新对HABI分子在受光照形成自由基后重新组合的过程进行了研究[24]。他们通过在核磁共振仪器中安装紫外激光器,利用1H-NMR和13C-NMR的二维谱更加直接地研究了HABI在光照形成自由基后重新复合后的过程。在室温下(298K),1-2’HABI在光 照后形 成自由基,其复合后的产物在253K下以4-4’连接,而在203K下则以2-2’连接。这些研究结果更加强力的验证了之前关于HABI分子结构的推论。
综上所述,我们发现HABI中两分子的三苯基咪唑之间的键连方式具有很大的“柔性”。在传统的HABI分子结构中,与光致变色直接相关的结构是1-2’的N—C连接方式。在Jiro Abe课题组设计的一系列分子中如果两个三苯基咪唑之间是正常的“头对头”或者桥联结构具有一定的刚性,则其主要是1-2’的N—C连接方式。而如果两个咪唑环是“头对尾”或者桥联结构具有较大的“柔性”则其连接方式主要由具体的分子结构以及热力学过程控制。
3 HABI作为分子开关的应用
最近十几年Jiro Abe课题组对HABI分子设计与修饰的研究工作使得HABI分子具备了实际应用的可能。我们就近两年该课题组及本课题组对于HABI的实际应用做简单的介绍。
3.1 全息成像
全息成像技术被誉为下一代3D电视显示技术而受到广泛的关注。然而全息成像技术之所以一直不能投入使用主要是由于不能找到能够跟上实时成像系统速度的大尺寸的光敏感材料。其中,光折变聚合物材料是非常重要的一种光敏感材料。在受到外界刺激后,它能够在数秒钟内回到初始态。然而光折变效应需要比较高的工作电压来记录光信息。如果用光致变色材料去替代光折变材料则不需要外加电压。同时,大尺寸的的光致变色材料可以通过将其分散到聚合物中然后用旋涂的方法实现。而pseudogem-bisDPI[2.2]PC类 HABI分子开关在受到紫外线照射后,室温下的热褪色的半衰期τ1/2仅仅为几十个微秒,完全能够替代光折变材料。据此他们成功地将HABI分子开关运用于全息成像[25]。文献中的 HABI分子开关的分子结构以及全息成像系统见图1 7。
在此基础上,他们继续研究了往主体材料聚甲基丙烯酸甲酯(PMMA)中添加增塑剂(磷酸三邻甲苯酯)对于光致变色材料的热褪色速率的影响[26],进一步阐述了将新型的HABI分子运用于全息成像的可行性。
3.2 超分辨成像
近期荧光分子开关的一个重要的应用就是超分辨成像。普通的光学显微镜由于本身原理的限制,无法分辨尺寸小于200nm的结构。而超分辨成像系统则能够克服普通光学显微镜这一“天生”的分辨极限,其分辨尺寸能够到几十个纳米。超分辨成像系统需要光致变色材料在变色的同时有荧光的“开”和“关”的过程,或由“强”到“弱”或者由“弱”到“强”的变化。因此,设计出具备有荧光分子开关性能的HABI分子开关,构建具有不同荧光开关性能的分子库,运用于超分辨系统具有巨大的应用前景。近几年,我们课题组主要的研究方向为设计出能够运用于超分辨成像的HABI分子开关。

图17 文献中的HABI分子化学结构以及光致变色化学反应式,全息成像的光学系统Molecular structure,photochromism and holographic imaging of pseudogem-bisDPI[2.2]PC

图18 Pseudogem-FPI-DPI[2.2]PC分子结构以及光致变色反应式;荧光显微镜下的光致变色图案(-180℃)Molecular structure,photochromism and optical pattern of Pseudogem-FPI-DPI[2.2]PC
由于HABI作为荧光分子开关的研究历史并不是很长,具有荧光分子开关性能的HABI分子开关就更少。2013年,Jiro Abe课题组报道了Pseudogem-FPI-DPI[2.2]PC分子(分子结构见图18)[27]。还是以对环芳烃为核,他们将一个咪唑环的4号位的苯环替换为荧光素基团得到了具有绿色荧光的HABI分子。和类似结构的HABI分子一样,它具有很快的热褪色性能,然而其荧光开关的“明态”(on state)和“暗态”(off state)之间的“荧光开关比”较低,同时由于分子开关变色太快,其荧光变色仅能在-180℃才能被明显的观察到(如图18)。从实际室温下的荧光成像来看是太快了,不利于应用。
AIE(Aggregation Induced Emission)现象自从2001年被唐本忠院士发现以来,立即引起了科学界的关注[28]。AIE的发现使化学家设计固态情况下具有强荧光的有机分子变得得心应手。据此我们课题组将典型的具有AIE效应的分子四苯基乙烯(TPE)与HABI分子直接偶联得到了TPEHABI(见图19)[29]。由于 TPE类分子的 AIE效应,TPE-HABI在固态下也具有较强的荧光。据此我们将其运用于超分辨成像系统,成功地将距离为90nm的两个TPE-HABI荧光基团分辨开。不过,由于荧光基团(TPE)与 HABI太近,TPE-HABI在受激发后产生的荧光马上被激发光刺激生成的三苯基咪唑自由基所吸收然后以非辐射跃迁的形式耗散掉,大大降低了其荧光量子产率(ΦF)。同时,由于采用传统非桥联的HABI分子结构,其有色态的褪色较慢也不利于实际的应用。
之后,通过对比研究了萘酰亚胺直接与HABI偶联和利用羟乙基哌嗪将HABI与荧光基团萘酰亚胺“隔离”两种分子设计模式,我们发现将HABI分子和荧光基团分开可以获得荧光量子产率较高的 HABI分子(ΦF=25%)(见图20)[30]。但由于NI-HABI分子仍采用传统非桥联的分子结构,其热褪色较慢,使得实际使用很困难。如何设计具有高的荧光量子产率以及合适的热褪色速率的HABI分子开关运用于超分辨成像,还需要大量的研究工作。
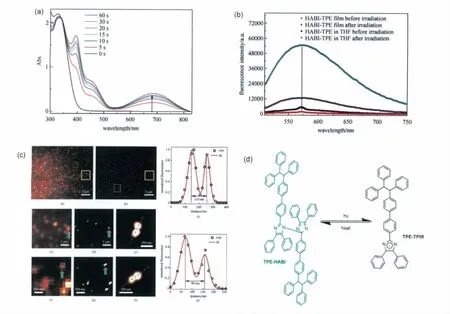
图19 (a)在365nm紫外光照射下TPE-HABI在苯中的吸收光谱;(b)固态以及四氢呋喃溶液中紫外线照射前后的发射光谱;(c)超分辨成像结果;(d)TPE-HABI的分子结构以及其光致变色的化学反应式(a)Absorption spectra of TPE-HABI upon 365nm irradiation in toluene;(b)fluorescence spectra of TPE-HABI upon 365nm irradiation in THF and solid state;(c)super-resolution fluorescence imaging;(d)molecular structure,photochromism of TPE-HABI
近期我们课题组已经开始着手设计Diffusion-Inhibited HABI分子,在引入荧光基团的同时,打断其与HABI的共轭连接从而在获得较快的热褪色速率下保持较强的荧光的量子产率的HABI分子,已取得较好的荧光开关性能,其在超分辨光学显微成像中的应用有望取得较大的进展(未发表的结果)。
4 HABI的氧化变质

图20 以萘酰亚胺为荧光基团与HABI非共轭连接的分子结构式,光致变色反应式以及光谱性质Molecular structure,photochromism and spectra of non-conjugated HABI-NI
衡量一个光致变色材料性能,一个重要参数就是其抗疲劳性。而与抗疲劳性相对应的就是光致变色材料的变质。HABI作为光致变色材料也不例外,在某些条件下也能发生变质。在此,我们仅仅通过已报道的研究工作来讨论HABI被氧化变质,至于HABI与活泼H反应生成三苯基咪唑的变质以及其它可能的变质途径,由于没有现成的研究工作,我们暂不列入讨论范围。
最早的关于HABI被氧化的报道是1962年Hayashi和Maede在研究lopine的氧化产生化学发光的研究,其中发生的主要的化学过程的机理如图21所示[31]。Hayashi和 Maede氧化的虽然不是HABI,但是这对于HABI后来的发现以及HABI的氧化机理起了基础作用。之后Guy Rio和Bernard Serkiz发表于Journal of the Chemical Society,Chemical Communication 的 论 文 提 出HABI被氧化主要的方式是1O2与咪唑环4,5位上的碳碳双键的加成作用[32]。近几年,Jiro Abe课题组对于HABI分子的改性使得HABI真正作为实用性的光致变色材料成为可能。HABI分子,尤其是Jiro Abe课题组改性后的桥联绑定(Diffusion Inhibited)HABI的抗氧化性如何也逐渐引起了关注。2012年,Jiro Abe课题组以1,8-bisTPI-naphthalene为对象研究了其与O2作用被氧化的过程,其氧化的过程见图22[33]。

图21 Lophine被氧化产生化学发光的反应机理Oxidation reaction and chemoluminescence mechanism of Lophine
由图22,我们可以看到在1,8-bisTPI-naphthalene分子中,O2分别与两个三苯基咪唑的5号位和4’位作用,桥联在一起,继而发生一系列的化学变化使其变质。之后,Andrew Beeby等选取传统的HABI分子研究了其被O2氧化的过程[34]。通过高分辨质谱以及对获得氧化产物的单晶X-射线衍射的分析,他们更加直接地证明了HABI可以被O2氧化,并指出HABI分子被氧化是一个普遍性的现象,即:无论是Jiro Abe课题组设计的Diffusion Inhibited HABI 还 是 传 统 的 HABI分子都可以被O2氧化。由此,HABI作为新型的分子开关在应用过程中被氧化的问题已摆上了化学工作者的桌面。如何进一步在化学结构上对HABI进行改性是未来HABI分子研究的重要内容。
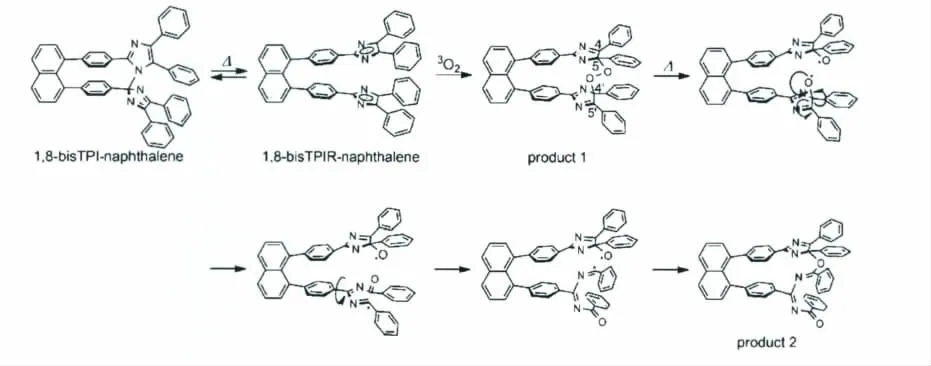
图22 1,8-bisTPI-naphthalene与O2作用被氧化的反应过程机理图Oxidation reaction mechanism of 1,8-bisTPI-naphthalene
5 展望
HABI分子自从1960年首次被报道以来,立刻引起了科学家的关注。然而由于传统HABI分子在结构上的缺陷,使得其作为光致变色材料方面的性能一直被“埋没”。随着近十几年(2001~2013)Jiro Abe课题组对HABI分子的改性,使得其热褪色半衰期由几分钟缩短至几十微秒,尤其是近期其在全息成像上的应用,使人们看到了HABI分子作为光致变色材料的巨大潜力。在超分辨成像技术的应用中,我们课题组刚刚起步,但是随着对HABI分子不断进行改性,合成出适用于超分辨成像的HABI荧光分子开关,有望大幅提升分子开关在超分辨光学显微成像领域的应用空间。与此同时,和其他所有的光致变色材料一样,对于其抗疲劳性的研究也会随着HABI分子的发展而更加深入。如何进一步对HABI分子的结构进行改性,从而从根本上提高其抗疲劳性也将是HABI分子设计的主题。
[1] 樊美公,姚建年,佟振合,等.分子光化学与光功能材料科学[M].北京:科学出版社,2009.Fan M G,Yao J N,Tung C H.Molecular Photochemistry and Optical Functional Material Sciences[M].Beijing:Science Press,2009.
[2] Bouas-Laurent H,Durr H.Organic photochromism[J].Pure and Applied Chemistry,2001,73(4):639-665.
[3] Fritzsche J.Note sur les carbures d′hydrog ne solides,tirés du gaudron de houille[J].Comptes rendus de l′Acad mie des Sciences,1867,69:1035-1037.
[4] Hayashi T,Maeda K,Shida S,Nakada K.A new phototropic substance and its ESR[J].Journal of Chemical Physics,1960,32:1568-1573.
[5] Hayashi T,Maeda K.Preparation of a new phototropic substance[J].Bulletin of the Chemical Society of Japan,1960,33(4):565-566.
[6] Zimmermann H,Baumg rtel,H,Bakke F.1,1′-bis-pyrryle,1,1′-bis-imidazyle und ihre dissoziation in radikale[J].Angewantle Chemie,1961,73(24):808-811.
[7] Ueda H.Electron spin resonance of some free radicals and related negative Ions[J].The Journal of Physical Chemistry,1964,68(6):1304-1310.
[8] Hayashi T,Maeda K,Takeuchi M.A.Kinetic study of the photochromism of 2,2′,4,4′,5,5′-hexaphenyl-1,1′-biimidazolyl with electron spin resonance[J].Bulletin of the Chemical Society of Japan,1964,27(11):1717-1718.
[9] Wilks M A J,Willis M R.Kinetics of the photochromic decay reaction of solutions of 2,2′,4,4′,5,5′hexaphenyl bi imidazole[J].Nature,1966,212:500-502.
[10] White D M,Sonnenberg J.Oxidation of triarylimidazoles:structures of the photochromic and piezochromic dimers of triarylimidazyl radicals[J].Journal of the American Chemical Society,1966,88(16):3825-3829.
[11] Cohen L R.Substituent effects on the reactivity of triarylimidazolyl free radicals toward tris(2-methyl-4-diethylaminophenyl)methane[J].Journal Organic Chemistry,1971,36(16):2280-2284.
[12] Tanino H,KondoT,Okada K,Goto T.Structures of three isomeric dimers of 2,4,5-triphenylimidazolyl[J].Bulletin of the Chemical Society of Japan,1972,45(5):1474-1480.
[13] Dessauer R.Photochemistry,History and Commercial Applications of Hexaarylbiimidazoles[M].Amsterdam,London:Elsevier,2006.
[14] Iwahori F,Hatano S,Abe J.Rational design of a new class of diffusion-inhibited HABI with fast back-reaction[J].Journal of Physical Organic Chemistry,2007,20(11):857-863.
[15] Hatano S,Abe J.Activation parameters for the recombination reaction of intramolecular radical pairs generated from the radical diffusion-inhibited HABI derivative[J].The Journal of Physical Chemistry A,2008,112(27):6098-6103.
[16] Fujita K,Hatano S,Kato D,Abe J.Photochromism of a radical diffusion-inhibited hexaarylbiimidazole derivative with intense coloration and fast decoloration performance[J].Organic Letters,2008,10(14):3105-3108.
[17] Kishimoto Y,Abe J.A fast photochromic molecule that colors only under UV light[J].Journal American Chemical Society,2009,131(12):4227-4229.
[18] Harada Y,Hatano S,Kimoto A,Abe J.Remarkable acceleration for back-reaction of a fast photochromic molecule[J].The Journal of Physical Chemistry Letters,2010,1(7):1112-1115.
[19] Zhou Y H,Baker W E,Kazmaier P M,Buncel E.Phenanthroimidazole dimers:structure and free radical reactivity;piezochromism,thermochromism,and photochromism[J].Canadian Journal of Chemistry,1998,76(6):884-895.
[20] Hatano S,Horino T,Tokita A,Oshima T,Abe J.Unusual negative photochromism via a short-lived imidazolyl radical of 1,1′-binaphthyl-bridged imidazole dimer[J].Journal of the American Chemical Society,2013,135(8):3164-3172.
[21] Mutoh K,Shima K,Yamaguchi T,Kobayashi M,Abe J.Photochromism of a naphthalene-bridged imidazole dimer constrained to the “anti”conformation[J].Organic Letters,2013,15(12):2938-2941.
[22] Kimoto A,Niitsu S,Iwahori F,Abe J.Formation of hexaarylbiimidazole heterodimers via the cross recombination of two lophyl radicals[J].New Journal of Chemistry,2009,33(6):1339-1342.
[23] Hatano S,Sakai K,Abe J.Unprecedented radical radical reaction of a[2.2]paracyclophane derivative containing an imidazolyl radical moiety[J].Organic Letters,2010,12(18):4152-4155.
[24] Delbaere S,Oriob M,Berthet J,Sliwa M,Hatano S,Abe J.Insights into the recombination of radical pair in hexaarylbiimidazoles[J].Chemical Communications,2013,49(52):5841-5843.
[25]Ishii N,Kato T,Abe J.A real-time dynamic holographic material using a fast photochromic molecule[J].Scientific Reports,2012,2:819.
[26] Ishii N,Abe J.Fast photochromism in polymer matrix with plasticizer and real-time dynamic holographic properties[J].Applied Physics Letters,2013,102:163301-163305.
[27] Mutoh K,Sliwa M,Abe J.Rapid fluorescence switching by using a fast photochromic[2.2]paracyclophane-bridged imidazole dimer[J].The Journal of Physical Chemistry C,2013,117(9):4808-4814.
[28] Luo J,Xie Z,Lam J.W.Y,Cheng L,Chen H,Qiu C,Kwok H.S,Zhan X,Liu Y,Zhu D,Tang B.Z.Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole[J].Chemical Communications,2001,18:1740-1741.
[29] Gong W L,Hu Z,Li C,Zhang G F,Chen T,Aldred M P,Huang Z L,Zhu M Q.Condensed state fluorescence switching of hexaarylbiimidazole-tetraphenylethene conjugate for super-resolution fluorescence nanolocalization[J].Frontiers of Optoelectronics,2013,6(4).DOI 10.1007/s12200-013-0330-1.
[30] Gong W L,Zhang G F,Li C,Aldred M P,Zhu M Q.Design,synthesis and optical properties of a green fluorescent photoswitchable hexaarylbiimidazole (HABI)with non-conjugated design[J].RSC Advances,2013,3(24):9167-9170.
[31] Hayashi T,Maeda K.Mechanism of chemiluminescence of 2,4,5-triphenylimidazole[J].Bulletin of the Chemical Society of Japan,1962,35:2057-2058.
[32] Rio G,Serkiz B.A photo-oxide of the 1,2′dimer of the 2,4,5-triphenylimidazolyl(Lophyl)radical[J].Journal of the Chemical Society,Chemical Communications,1975,20:849-850.
[33] Hatano S,Abe J.A peroxide-bridged imidazole dimer formed from a photochromic naphthalene-bridged imidazole dimer[J].Physical Chemistry Chemical Physics,2012,14(16):5855-5860.
[34] Edkins R M,Probert M R,Fucke K,Robertson C M,Howard J A K,Beeby A.The formation of peroxide degradation products of photochromic triphenylimidazolyl radicaldimers[J].Physical Chemistry Chemical Physics,2013,15(20):7848-7853.
