Aurora-A激酶及其抑制剂研究进展
2014-02-16许晓辉孙陶利朱玉婷
许晓辉,孙陶利,朱玉婷
Aurora-A激酶及其抑制剂研究进展
许晓辉,孙陶利,朱玉婷
Aurora-A是Aurora激酶家族中的重要成员之一,在细胞有丝分裂过程中发挥着重要功能,Aurora-A的过度表达与人类多种恶性肿瘤紧密相关。鉴于Aurora-A的重要作用,其被作为一个设计抗肿瘤药物的新靶点。目前已有几种选择性Aurora-A激酶抑制剂进入到临床研究中,并且表现出良好的治疗效果。本文从Aurora-A激酶的生物学、与肿瘤的密切关系及其抑制剂的研发状况进行综述。
Aurora-A;有丝分裂;抗肿瘤;激酶抑制剂
近几年来,人们发现一种新的能调节中心体和微管功能的丝氨酸-苏氨酸蛋白激酶-Aurora蛋白家族,Aurora激酶的突变会导致中心体分离异常,形成单极纺锤体。Aurora-A是Aurora激酶家族中最重要的成员,是有丝分裂过程中最主要的调节器,与人类多种恶性肿瘤紧密相关。Aurora-A激酶独特的药理作用机制以及与恶性肿瘤的关系,使得它成为抗肿瘤药物研究的重要靶点,而其抑制剂也被认为是具有良好开发前景的新型抗肿瘤药物。
1 Aurora激酶概述
Aurora激酶是中心体相关激酶,存在于真核细胞内,在中心体复制、两极纺锤体形成、染色体重排和染色体检查点监测等重要的有丝分裂过程中发挥着至关重要的作用[1]。Aurora激酶由3个亚群组成,即Aurora-A、Aurora-B和Aurora-C。这3个亚群的蛋白质的一级结构均含有一个N-端调控区和一个C-端催化区,且催化区序列相似度达71%,具有高度的保守性,然而它们三者的N末端却不相同[2](图1)。这种催化区结构的保守性表明Aurora激酶的底物在结构上具有特异性,而Aurora激酶异常表达则可能导致遗传不稳定或者诱发肿瘤的增长。
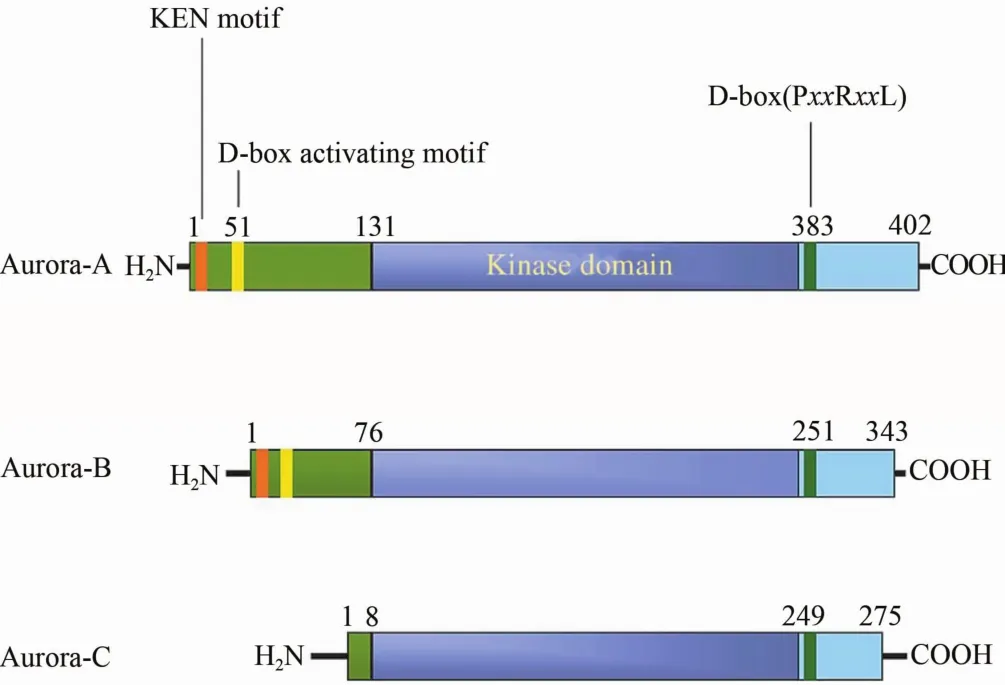
图1 Aurora-A、Aurora-B和Aurora-C的结构
2 Aurora-A激酶生物学
2.1 Aurora-A激酶结构 Aurora-A激酶的编码基因定位于人第20号染色体20q13.2,全长cDNA包含1 209 kb的开放阅读框,有9个外显子,编码一种相对分子质量为46×103、403个氨基酸组成的丝氨酸和苏氨酸激酶[3]。Aurora-A激酶具有一个高度保守的C末端催化结构域,其由一个N端β链结构域和一个C端α螺旋结构域组成。另外,C端的催化结构域中包括2个保守的序列即活化环和降解盒。Aurora-A的N端是定位结构域,它的功能是将Aurora-A以依赖微管的方式定位于中心体上[4]。
晶体结构研究表明,Aurora激酶的活性位点可以划分成4个部分[5]:①溶剂暴露前口袋区;②铰链区;③疏水的后口袋区;④磷酸盐结合区域。结构分析研究表明,溶剂暴露前口袋区和磷酸盐结合区都在ATP键合口袋的外部,并且高度暴露在溶剂中,因此可以根据这两个区域的特性来对候选药物进行结构修饰,从而改善其药代动力学性质。
2.2 Aurora-A激酶的功能与激活 有研究表明,Aurora-A的mRNA、蛋白水平、激酶活性及在细胞内的分布,其动态随细胞周期各时相的转换而改变。Aurora-A在细胞分裂的前期主要定位于中心体周围,中期在纺锤体极附近的微管上,后期和末期位于极性微管上,主要负责中心体的繁殖和分离,双极纺锤体的聚集,以及有丝分裂进入和退出,对中心体的成熟和纺锤体的装配起着重要作用[6]。Aurora-A激酶的扩增和过度表达帮助创造了遗传改变所必要的条件从而使肿瘤发生。
Aurora-A激酶通过与其底物结合,发生自身磷酸化从而被激活。被激活后的Aurora-A可以防止其被激酶相关联的Ⅰ型磷酸酶去磷酸化[7]。Aurora-A的底物包括TPX2、Ajuba、EG5、CDC25B、p53和BRCA-1等[8]。激活Aurora-A激酶的底物和Aurora-A激酶抑制剂之间的平衡对正常的有丝分裂是非常重要的。因此,提高以及降低Aurora-A激酶的活性都可能会导致错误的有丝分裂。
总结Aurora-A激酶在有丝分裂中的功能和其相应底物与其作用的结果,如图2所示。

图2 Aurora-A的功能和底物的相互作用
3 Aurora-A激酶与恶性肿瘤
Aurora-A激酶的编码基因,定位在20q13.2,该区段染色体具有易位、缺失或扩增活跃的特点[3],表明它具有天然的不稳定性。这些研究表明,当Aurora-A过度表达时,它是一种潜在的致癌基因。Aurora-A的过度表达可能是由于扩增的基因,诱导基因的转录及翻译后的不稳定性引起的[9]。研究发现,Aurora-A普遍存在于人类恶性肿瘤中并有扩增的现象,如结肠癌、胰腺癌、卵巢癌、胃癌、乳腺癌以及甲状腺癌等[10]。
4 Aurora-A激酶抑制剂
作为一个重要的抗癌药物新靶标,Aurora激酶已经吸引了很多制药公司和科研机构对其抑制剂进行研究、开发和结构改造。目前,已经有大量关于Aurora激酶小分子抑制剂的报道,其作用机制主要是通过竞争性的作用于疏水性的ATP结合口袋从而发挥抑制作用[5]。总结目前已报道的Aurora激酶抑制剂,其中大部分抑制剂属于Aurora激酶多重抑制剂,鉴于多重抑制剂存在较大的不良反应,使得发现高效、低毒的选择性抑制剂成为当前Aurora激酶类药物研究的必然趋势。
目前,选择性Aurora激酶抑制剂的研究主要是针对Aurora-B激酶的,针对Aurora-A激酶的选择性抑制剂则很少。但相对于Aurora-B而言,Aurora-A在结肠癌、卵巢癌、胃癌以及乳腺癌等许多肿瘤中均过量表达[10],并且Aurora-A激酶的过度表达使细胞有丝分裂检测点的监测功能失控,导致有丝分裂异常,癌基因扩增,正常细胞转化为癌细胞[11]。由此可见,Aurora-A能更加有效地引起细胞有丝分裂停滞并快速诱导细胞凋亡[12],因而以Aurora-A为靶点的抗肿瘤药物治疗在一定程度上将优于选择性Aurora-B激酶抑制剂。
4.1 选择性Aurora-A激酶抑制剂
4.1.1 MLN8054 MLN8054(Millennium,图3A,化合物1)是第一个口服有效并且具有选择性的苯并氮杂类Aurora-A激酶抑制剂,对Aurora-A和urora-B的半抑制浓度(50%inhibitory concentration,IC50)分别为4 nmol/L和172 nmol/L,对Aurora-A的选择性比Aurora-B高出40倍左右[13],属于ATP竞争性可逆抑制剂。MLN8054对人类结肠癌、前列腺癌和非小细胞肺癌均表现出抗肿瘤活性。在体内模型中,用低浓度的MLN8054抑制Aurora-A会导致有丝分裂纺锤体形成异常,而高浓度的MLN8054会造成组蛋白H3的磷酸化缺失。在Ⅰ期临床研究中,MLN8054以口服给药,能被快速吸收,消除半衰期大约为35 h。MLN8054的耐受性良好,大剂量给药没出现骨髓抑制和黏膜毒性,但出现了嗜睡的不良反应。近期MLN8054的2个Ⅰ期临床研究都已被中止。
4.1.2 MLN8237 MLN8237(图3B,化合物2)是由MLN8054改良而来的第二代Aurora-A选择性抑制剂。在体外酶活性筛选中,MLN8237对Aurora-A的IC50为1 nmol/L,比对Aurora-B的抑制效果强200倍。MLN8237能抑制多种癌细胞的增殖,如HCT-116、PC3、SK-OV-3和LY-3等。MLN8237和尼罗替尼的联合治疗还能增强尼罗替尼的抗肿瘤效果,MLN8237和多西他赛的联合治疗也能增强多西他赛的抗套细胞淋巴瘤的效果[14],Ⅰ期临床主要用于实体肿瘤以及血液瘤的治疗。
4.1.3 ENMD-2076 ENMD-2076(图3C,化合物3)是一个口服有效的乙烯嘧啶碱类化合物,选择性的抑制Aurora-A,对Aurora-A和Aurora-B抑制活性的IC50分别为14 nmol/L和290 nmol/L。研究表明,ENMD-2076能使细胞周期停滞在G2/M期,最终导致细胞凋亡[15]。动物实验研究显示,ENMD-2076具有抗血管生成、阻碍细胞周期以及抗增殖活性,这为该化合物用于治疗包括多发性骨髓瘤在内的血液癌提供了支持。ENMD-2076的Ⅰ期临床研究始于2008年3月,用于治疗实体肿瘤,目前已经制定了治疗血液瘤的Ⅰ期临床研究计划。
4.1.4 其他 Aliagas-Martin报道[16]了一种嘧啶类的Aurora-A激酶的选择性抑制剂(图3D,化合物4),它抑制Aurora-A、Aurora-B的IC50分别为34 nmol/L、34 000 nmol/L,对Aurora-A的选择性是Aurora-B的1 000倍。此化合物虽然对Aurora-A的选择性很高,但是抗肿瘤细胞增殖的能力却不是特别强,可能与其自身较差的透膜能力有关。
另外,挪威学者Prime等[17]报道的一类新型吡唑取代的酞嗪酮类化合物(图3E,化合物5),对Aurora-A激酶抑制的选择性大于1 000,目前是否进入临床前研究尚不清楚。
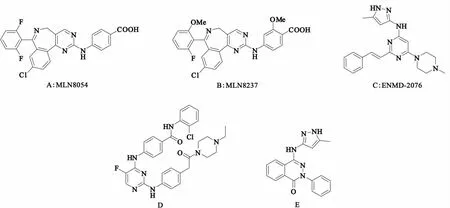
图3 化合物1~5结构式
4.2 Aurora-A、Aurora-B和Aurora-C多重激酶抑制剂
4.2.1 MK0457(VX-680) MK0457(VX-680,图4A,化合物6)是靶向ATP结合位点的嘧啶类Aurora激酶抑制剂,对3类Aurora激酶均有抑制作用,抑制Aurora-A、Aurora-B和Aurora-C的IC50分别为0.6、18、4.6 nmol/L[18]。VX-680在Ⅰ期临床试验中主要用于治疗慢性髓细胞白血病以及T315IBcr-Abl突变的急性淋巴细胞性白血病。由于可能引起的QTc间期延长风险,VX-680的Ⅱ期临床并未进行。
4.2.2 ZM447439 ZM447439(图4B,化合物7)是一种ATP竞争性Aurora激酶抑制剂,是喹唑啉衍生物对Aurora-A和Aurora-B的抑制IC50分别为110 nmol/L、130 nmol/L[19]。ZM447439能调控细胞中的染色体排列、细胞分裂和有丝分裂检查点,但不影响许多基本的细胞周期调节因子的活动,如CDC25、 MAPK、cyclinB等。目前,ZM447439已进入Ⅰ期临床研究阶段。
4.2.3 PHA-739358 PHA-739358(图4C,化合物8)是一个吡咯并吡唑类的ATP竞争性抑制剂,对Aurora-A、Aurora-B、Aurora-C的抑制IC50分别为13、79、61 nmol/L[20]。临床前研究中PHA-739358能够诱导肿瘤细胞多倍性的产生。目前PHA-739358已经进入Ⅱ期临床研究,用于治疗伊马替尼耐药的慢性粒细胞白血病以及靶向治疗服用c-Abl抑制剂之后癌细胞转移的前列腺癌。
4.2.4 其他 对Aurora激酶有多重抑制效果的化合物还有SNS-314[21]、AT-9283[22]、CCT137690[23]、AC081[24]等,它们对Aurora-A都有很明显的抑制效果,并且也都进入到临床研究阶段,有望寻找出更加高效低毒的Aurora-A激酶抑制剂。
4.3 Aurora-A激酶抑制剂的结构修饰 在寻找高效低毒的选择性Aurora-A激酶抑制剂过程中,有很多化合物会因为临床前研究或临床试验阶段出现某种缺点而失去进一步研究的价值,比如MLN8054引起了嗜睡的不良反应,临床研究已被终止;化合物4虽然对Aurora-A的选择性是Aurora-B的1 000倍,但由于自身的透膜能力低而导致了其抗肿瘤细胞增值能力弱,阻碍了它的成药性。
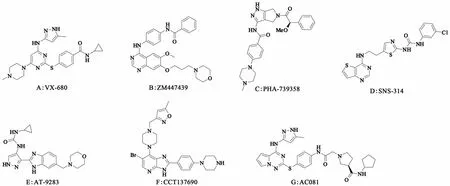
图4 化合物6~12结构式
为了更快更好的发现新型的选择性Aurora-A激酶抑制剂,在已知化合物结构的基础上,对其进行进一步的结构修饰,使其成药性增加不失为一条发现新药的途径。进行化合物结构优化时,主要可以从以下两个大方面进行:①保留具有选择性的母核结构,使化合物在保持对Aurora-A激酶高度选择性的基础上,引入其他取代基团,改变化合物的性质;②发现新的母核结构,提高化合物对Aurora-A激酶的选择性。
在结构修饰的第一方面中,通过引入新的取代基团,可以改善化合物的以下性质:①提高化合物的抑酶活性;②改善化合物的溶解性;③提高化合物的透膜能力;④提高化合物的稳定性;⑤改善化合物药代动力学性质;⑥降低化合物的不良反应。
5 展望
目前已进入临床研究阶段的Aurora激酶多重抑制剂,有很多都引起了不同程度的不良反应,不利于进一步的研究。而随着Aurora-A激酶生物结构和功能研究的逐渐深入,研究者们对选择性Aurora-A激酶抑制剂的研发也有了很大的进步。这些具有选择性的Aurora-A激酶抑制剂具有靶向治疗的作用,可以有效的减少临床应用阶段出现的不良反应。因此,开发更加高效低毒的选择性Aurora-A激酶抑制剂,必将为研发新型抗肿瘤药物提供一条新的思路,也可为肿瘤的靶向治疗作出一定的贡献。
[1]Liu Q,Ruderman JV.Aurora A,mitotic entry,and spindle bipolarity[J].Proc Natl Acad Sci USA,2006,103(15):5811-5816.
[2]Katayama H,Sen S.Aurora kinase inhibitors as anticancer molecules[J].Biochim Biophys Acta,2010,1799(10/12):829-839.
[3]Fu J,Bian M,Jiang Q,etal.Roles of Aurora kinases inmitosis and tumorigenesis[J].Mol Cancer Res,2007,5(1):1-10.
[4]Mountzios G,Terpos E,Dimopoulos MA.Aurora kinases as targets for cancer therapy[J].Cancer Treat Rev,2008,34(2):175-182.
[5]Vulpetti A,Bosotti R.Sequence and structural analysis of kinase ATP pocket residues[J].Farmaco,2004,59(10):759-765.
[6]Marumoto T,Zhang D,Saya H.Aurora-A--a guardian of poles[J].Nat Rev Cancer,2005,5(1):42-50.
[7]Karthigeyan D,Prasad SB,Shandilya J,et al.Biology of Aurora A kinase:implications in cancer manifestation and therapy[J].Med Res Rev,2011,31(5):757-793.
[8]Jackson JR,Patrick DR,Dar MM,et al.Targeted anti-mitotic therapies,can we improve on tubulin agents[J].Nat Rev Cancer,2007,7(2):107-117.
[9]Andrews P.Aurora kinases,shining lights on the therapeutic horizon[J].Oncogene,2005,24(32):5005-5015.
[10]Green MR,Woolery JE,Mahadevan D.Update on aurora kinase targeted therapeutics in oncology[J].Expert Opin Drug Discov,2011,6(3):291-307.
[11]Foote KM,Mortlock AA,Heron NM,et al.Synthesis and SAR of 1-acetanilide-4-aminopyrazole-substituted quinazolines:selective inhibitors of Aurora B kinase with potent anti-tumor activity[J].Bioorg Med Chem Lett,2008,18(6):1904-1909.
[12]Barr AR,Gergely F.Aurora-A:the maker and breaker of spindle poles[J].JCell Sci,2007,120(Pt17):2987-2996.
[13]Manfredi MG,Ecsedy JA,Meetze KA,et al.Antitu-mor activity of MLN8054,an orally active small-molecule inhibitor of Aurora A kinase[J].Proc Natl Acad Sci USA,2007,104(10):4106-4111.
[14]QiW,Cooke LS,Liu X,et al.Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity inmantle cell lymphoma[J].Biochemical Pharmacology,2011,81(7):881-890.
[15]Wang S,Midgley CA,Scaërou F,etal.Discovery of N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors[J].JMed Chem,2010,53(11):4367-4378.
[16]Aliagas-Martin I,Burdick D,Corson L,et al.A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually High selectivity against Aurora B[J].JMed Chem,2009,52(10):3300-3307.
[17]Prime ME,Courtney SM,Brookfield FA,et al.Phthalazinone pyrazoles as potent,selective,and orally bioavailable inhibitors of Aurora-A kinase[J].JMed Chem,2011,54(1):312-319.
[18]Tyler RK,Shpiro N,Marquez R,et al.VX-680 inhibits Aurora A and Aurora B kinase activity in human cells[J]. Cell Cycle,2007,6(22):2846-2854.
[19]Li M,Jung A,Ganswindt U,et al.Aurora kinase inhibitor ZM447439 induces apoptosis via mitochondrial pathways[J].Biochem Pharmacol,2010,79(2):122-129.
[20]Arpinelli P,Ceruti R,Giorgini ML,et al.PHA-739358,a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer[J].Mol Cancer Ther,2007,6(12 Pt1):3158-3168.
[21]Arbitrario JP,Belmont BJ,Evanchik MJ,et al.SNS-314,a pan-Aurora kinase inhibitor,shows potent antitumor activity and dosing flexibility in vivo[J].Cancer Chemother Pharmacol,2010,65(4):707-717.
[22]Kimura S.AT-9283,a small-moleculemulti-targeted kinase inhibitor for the potential treatment of cancer[J].Curr Opin Investig Drugs,2010,11(12):1442-1449.
[23]Cee VJ,Schenkel LB,Hodous BL,et al.Discovery of a potent,selective,and orally bioavailable pyridinyl-pyrimidine phthalazine aurora kinase inhibitor[J].J Med Chem,2010,53(17):6368-6377.
[24]彭文,张小猛,张仓,等.靶向非活性激酶DFG-out变构结合位点的研究进展[J].中国新药杂志,2012,1(8):890-894.
The research progress of a Aurora-A kinase and its inhibitors
XU Xiaohui1,SUN Taoli2,ZHU Yuting3
(1.Pingliang Centre for Drug Inspection,Pingliang Gansu 744000,China;2.The Basical Department of Medicine,Xiangnan University,Binzhou Hunan 423000,China;3.Department of Pharmacy,People′s Hospital of Hanzhong,Hanzhong Shanxi723000,China)
Aurora-A is an important member of Aurora kinase family,which plays a significant function in cell mitosis process.The over expression of Aurora-A is closely associated with a variety of human tumors.Aurora-A is designed as a new target for anticancer drugs,owing to itsmajor function in cell mitosis.At present,a few selective inhibitors of Aurora-A kinase have entered the clinical study and showed good therapeutic effect.In this article,we will state the biological properties of Aurora-A kinase.At the same time,we will introduce the close relationship between Aurora-A kinase and tumor,and further outline the development status of Aurora-A kinase inhibitors.
Aurora-A;Mitosis;Antitumor;Kinase inhibitors
R284.1;R914.5
A
2095-3097(2014)02-0079-05
10.3969/j.issn.2095-3097.2014.02.004
2014-02-25 本文编辑:冯 博)
744000甘肃平凉,平凉市药品检验检测中心(许晓辉);423000湖南郴州,湘南学院基础医学部(孙陶利);723000陕西汉中,汉中市人民医院药剂科(朱玉婷)
