某1000MW燃煤机组脱硝催化剂运行性能检测
2014-02-07赵冬梅蒋惠铮陆金丰肖雨亭江苏龙源催化剂有限公司江苏无锡245国电集团脱硝催化剂分析测试中心江苏无锡245
赵冬梅,蒋惠铮,贾 曼,陆金丰,肖雨亭(. 江苏龙源催化剂有限公司,江苏 无锡 245;2. 国电集团脱硝催化剂分析测试中心,江苏 无锡 245)
0 引言
SCR烟气脱硝技术是应用较多、较为成熟的一种技术,催化剂是SCR脱硝技术的核心。目前,燃煤电厂脱硝系统应用最广泛的催化剂是V2O5-WO3-TiO2体系的SCR催化剂,国内外学者对该体系催化剂性能衰减趋势、中毒失活规律做过许多研究[1-3]。但是,这些研究往往基于实验室某种特定条件下进行,不能完全反应催化剂实际运行的情况。我国火电机组燃煤复杂,煤种多变,装机时间较早的一批催化剂已连续运行超过24000h甚至更多,有必要对该体系的催化剂在国内燃煤条件下的实际运行性能、活性衰减等情况进行研究。
本文选取了某1000MW机组使用的脱硝催化剂,分别对未使用的新鲜催化剂、使用约8000h和24000h的催化剂做了跟踪检测,通过对实际运行的催化剂性能检测对比,对催化剂的运行维护提供一定的借鉴。
1 试验部分
1.1 分析仪器
BET比表面积采用SA3100TM比表面及孔隙分析仪(贝克曼库尔特有限公司)测定;微孔体积采用PoreMaaster-33压汞仪(美国康塔仪器公司)测定;硫酸根采用CS-800硫分析仪(德国埃尔特公司)测定;中毒元素采用ICP-OES-715电感耦合等离子发射光谱仪(美国瓦里安技术有限公司)测定;抗压强度采用压力试验机测定;磨损强度采用磨损强度模拟试验装置测定(江苏龙源催化剂有限公司自制);脱硝活性采用微型活性测试装置测定(江苏龙源催化剂有限公司自制)。
1.2 分析方法
催化剂分析方法主要按照相关标准进行[4]。中毒元素含量分析:采用高浓度混合酸对催化剂进行加热至完全消解,使用电感耦合等离子发射光谱仪(ICP)分析样品中主要中毒元素Fe、K、Na含量。
采用硫分析仪分析催化剂粉末的硫含量。采用比表面及孔隙分析仪对催化剂粉末样品作比表面积分析。催化剂孔容分析采用压汞仪对催化剂粉末样品进行分析。同时,对样品进行抗压强度分析和磨损强度分析。脱硝活性分析:取催化剂样品3×3共9孔,长度30cm,使用微型活性测试装置对其进行脱硝活性测试 。
2 结果与讨论
2.1 催化剂表面沉积物和中毒元素分析
催化剂在长期的运行过程中,燃煤产生的飞灰会与催化剂长期接触,其中的碱金属、重金属及含硫化合物会逐渐沉积在催化剂表面,并与催化剂活性中心发生不可逆反应,导致催化剂活性中心的丧失和活性点位的减少。该催化剂运行后表面沉积物和中毒元素的分析结果见表1。
表1 催化剂主要中毒元素及硫酸根分析结果

项 目K/uL·L-1Na/uL·L-1Fe/uL·L-1硫酸根/%新鲜催化剂2192801541.8使用8000h4176022801.6使用24000h4257184612.0
从表1可以看出,随着运行时间的延长,催化剂中主要的碱金属含量逐渐增加。其中,与新鲜催化剂相比,在最初运行的8000h,K元素增加了90%以上,Na元素累积了150%以上,但是这两种元素在催化剂表面累积到一定程度后就趋于平缓了。这可能是因为,当燃用煤质趋于稳定后,活性位与这两种碱金属的反应趋于稳定,其在催化剂表面的作用趋于饱和。另一方面,与新鲜催化剂相比,Fe元素增加了近200%,并呈现出递增的趋势。这可能是因为,催化剂活性位与Fe元素的反应活性要高于K、Na元素,其在催化剂表面的作用还未饱和。对比三种碱金属的沉积趋势,可以发现:
(1)该1000MW机组催化剂没有明显的中毒现象,表面碱金属沉积符合正常的堕化趋势,碱金属沉积量在可控范围内。
(2)在催化剂运行的初期,碱金属迅速在催化剂表面沉积并逐渐饱和,说明催化剂中毒往往发生在运行的早期阶段。该分析结果的理论指导意义是,在运行的早期,燃用的煤质要尽量稳定,并且不要过多的偏离设计煤种,否则会过早导致催化剂中毒失活。
硫酸根的分析结果显示,在整个运行期内,催化剂表面的硫化合物沉积不明显,说明运行期间没有长期低温运行导致硫酸氢铵等硫酸盐的过多沉积,所燃用的煤种含硫量不高且比较稳定[5]。
2.2 BET比表面积分析
催化剂比表面积的测定结果见表2。从表2可知,与新鲜催化剂相比,使用8000h后催化剂的比表面积变小。
这可能是因为碱金属及其化合物占据了活性位并堵塞了部分微孔所致。单位体积(或质量)催化剂的活性主要取决于催化剂表面积的大小,因此,从比表面积的试验数据初步可知,运行催化剂的活性已经部分丧失。
表2 催化剂比表面积分析结果

项 目BET比表面积/m2·g-1新鲜催化剂58.0使用8000h51.4使用24000h46.5
2.3 微孔孔容及孔径分布分析
表3是催化剂样品的微孔总孔容和孔径分析结果。从表3可以看出,使用后催化剂的孔容逐渐降低,24000h后的降低幅度为12.5%,说明催化剂的孔道部分被堵塞。从平均孔径来看,使用后催化剂的平均孔径增大,由14.97nm增大到22.19nm,说明是催化剂的14nm以下的孔道出现了较多的堵塞情况。根据孔径分布结果分析,5~10nm的孔径分布明显减少,说明5~10nm孔径的细小微孔堵塞比较严重。同时,大于50nm的孔径分布增加了近50%。可能的原因:一是催化剂微孔结构受到烟气中细微粒子及部分化学沉积物的堵塞,导致细小微孔分布明显减少;二是催化剂在长期的高温运行过程中,载体部分发生了烧结作用,从而导致形成了新的大孔径微孔,而细小微孔进一步烧结堵塞甚至消失。上述两个原因共同作用的结果就是催化剂原有的微观结构遭到一定程度的破坏。
根据选择性催化还原的原理可知,烟气向催化剂表面和微孔内的外扩散是整个脱硝反应过程中速度最慢的步骤,因而也是脱硝反应的控制步骤,而催化剂的微观结构会直接影响外扩散速率,从而影响催化剂的活性[6]。催化剂生产厂商往往通过配方和烧结工艺的优化,结合化学寿命、机械强度的要求,将催化剂的微观结构控制在平衡可控范围内,微观结构的改变,会或多或少的影响催化剂机械强度和化学寿命。微观结构劣化程度不大的催化剂,可以通过再生手段得到一定程度的恢复,但是过度烧结等因素引起的微观结构极端劣化的催化剂,目前尚无有效手段恢复其性能。
表3 催化剂微孔孔容及孔径分布分析结果
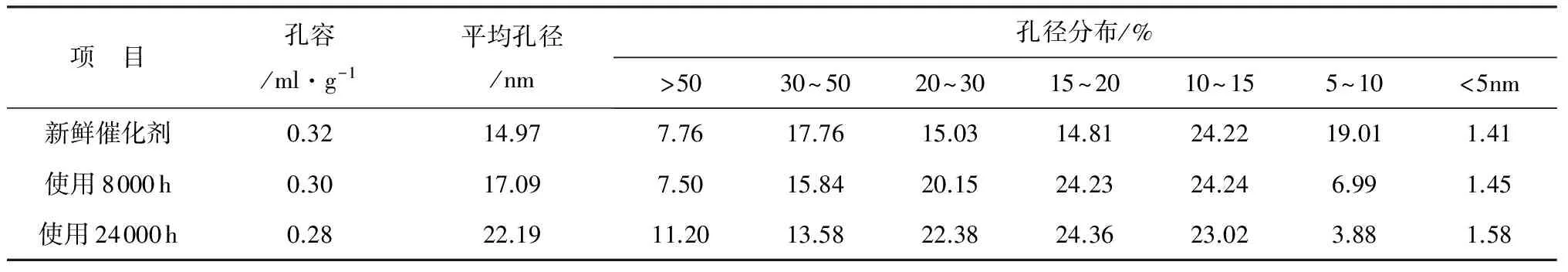
项 目孔容/ml·g-1平均孔径/nm孔径分布/%>5030~5020~3015~2010~155~10<5nm新鲜催化剂0.3214.977.7617.7615.0314.8124.2219.011.41使用8000h0.3017.097.5015.8420.1524.2324.246.991.45使用24000h0.2822.1911.2013.5822.3824.3623.023.881.58
2.4 机械强度分析
样品制备过程中,催化剂表面和内部在裁切过程中发现多处裂纹,有些裂纹为陈旧性裂纹,应是催化剂制造过程中产生,有些经判断应是运行中出现的,新出现的裂纹说明催化剂的机械强度已开始出现下降趋势(见表4)。从表4中可以看出,催化剂抗压强度和磨损强度整体呈现逐渐下降的趋势,但下降的程度不大。分析结果表明,经过一个化学寿命周期的运行,催化剂磨损强度和抗压强度虽然有下降趋势,但是未明显劣化,属于催化剂正常的坠化现象,整体机械性能保持良好。
表4 催化剂机械强度分析结果
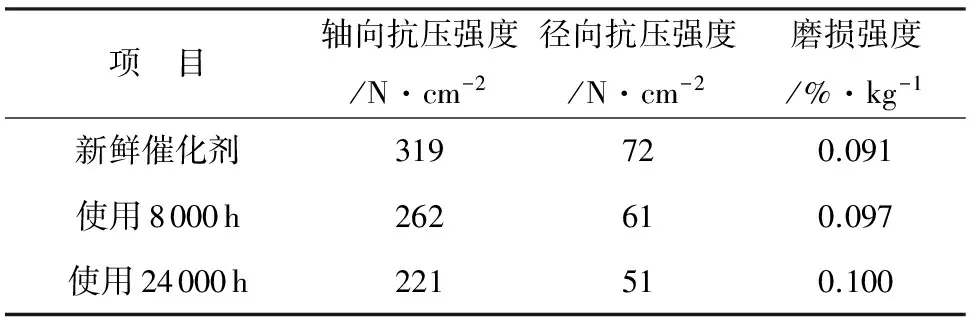
项 目轴向抗压强度/N·cm-2径向抗压强度/N·cm-2磨损强度/%·kg-1新鲜催化剂319720.091使用8000h262610.097使用24000h221510.100
2.5 催化活性分析
催化剂的活性分析结果见表5。
表5 催化活性分析结果
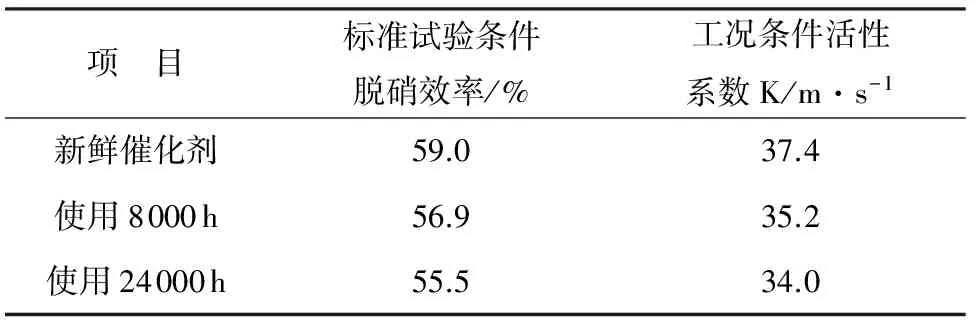
项 目标准试验条件脱硝效率/%工况条件活性系数K/m·s-1新鲜催化剂59.037.4使用8000h56.935.2使用24000h55.534.0
从表5可知,使用后催化剂活性呈逐渐下降的趋势,但是仍属于正常使用条件下的活性衰减。该分析结果表明,经过24000h的运行,该1000MW机组催化剂活性没有发生明显劣化,仍可继续服役使用,或添加加装层后满足更高的脱硝要求。
3 结语
以某1000MW燃煤机组脱硝系统使用的脱硝催化剂为研究对象,分别对不同运行时间的催化剂进行了对比检测。检测结果表明,运行初期,催化剂表面沉积了一定量的碱金属,但属于正常范围内。没有出现过多的硫化合物的沉积。催化剂微观结构遭到一定程度的破坏,比表面积有下降趋势。催化剂机械强度基本保持良好。催化剂活性衰减在正常范围内,催化剂可以继续使用或添加加装层。
[1]云 端,宋 蔷,姚 强. V2O5-WO3-TiO2系SCR催化剂失活机理及分析[J].煤炭转化,2009,(1): 91-96.
[2]Zheng Y J,Jensen A D,Johnsson JE, et al. Deactivation of V2O5-WO3-TiO2SCR catalyst at biomass fired power plants [J].Applied Catalysis B: Environmental,2008,(83):186-194.
[3]Lee B W, Cho H,Shin D W. Characterization and De-NOXactivity of binary V2O5/TiO2and WO3/TiO2, and ternary V2O5-WO3/TiO2SCR catalysts [J].Ceramic Processing Research,2007,(8):203-207.
[4]DL/T1286-2013,火电厂烟气脱硝催化剂检测技术规范[S].
[5]肖雨亭,陆金丰.脱硝催化剂的影响因素及选型[J].节能与环保,2012,(10):48-50.
[6]Parvulescu V I,Grange P,Delmon B. Catalytic removal of NO [J].Catalysis Today,1998,(46):233-316.
