静电作用力算法对分子动力学模拟结果的影响分析
2014-02-06李浩,白姝
李 浩,白 姝
(天津大学化工学院,天津 300072;天津大学系统生物工程教育部重点实验室,天津 300072)
分子模拟是基于原子的结构、势能等信息,利用统计学原理对系统宏观性质进行预测的一种非量子化计算方法,它具有足够小的时间和空间尺度,能让研究者方便地解析动力学过程的微观信息。然而,由于在处理作用力时所用的算法都只是近似处理,不能完全真实的描述作用力,因此非自然模拟结果时有发生[1-3]。近期,Wang等[4]研究发现同电荷交换介质可以显著提高带同种电荷的蛋白质复性收率,在研究中他们没有检测到蛋白质被吸附到介质表面,因此推测蛋白质和平板之间的作用力为静电排斥作用。但是,他们并没有从实验的角度考察此过程的机理。而在利用分子模拟研究此课题时发现,在实验所用的体系中,蛋白质和色谱基质间相互作用力类型很大程度上取决于所选取的力场参数,因此本研究就这个现象及其产生的原因进行了分析。
分子动力学模拟用于考察蛋白质之间[5]以及蛋白质和界面之间的相互作用已有很多报道。在以溶菌酶为目标蛋白的研究中,也有利用不同的模拟方法研究其在带电表面的行为[6-8]。但是,对于蛋白质在由带同种电荷的离子交换介质介导的静电作用过程鲜有研究者关注。同时,对于静电作用力的处理大部分采用的Cut-off算法,未考虑远程静电力可能对模拟结果带来的影响。
因此,本研究首先利用前期构建的静电作用模型来模拟带同电荷的离子交换介质。通过分子动力学模拟,考察不同的静电参数对蛋白质和带电表面作用过程的影响,展示了蛋白质在带电表面的行为,计算了此过程的构象和能量变化,初步揭示了溶菌酶体系下静电作用力参数的选取对分子动力学模拟的影响,揭示了蛋白质在界面行为的微观机理。为进一步研究蛋白质在界面表面的行为奠定了一定的理论基础。
1 模拟方法
1.1 模拟体系构建
根据蛋白质数据库(http://www.rcsb.org/pdb/)中的晶体衍射结构(PDB ID:3LYZ)构建溶菌酶的全原子模型[9],如图1a)所示。在pH值为7的条件下,溶菌酶带8个正电荷。由于实验中所用色谱介质孔道较为复杂,为节省计算量,前人将其简化为两互相平行的平板,包括模型化的带电表面以及惰性平板,平板之间距离为12nm,如图1b)所示。色谱介质由平板及配基构成,其中色谱介质的拓扑结构由 Dundee PRODRG 2.5 server(beta)[10]生成。根据实验情况,每个色谱配基带电量设置为1个正电荷,而对于模拟中配基的带电基团分配及每个基团的带电量通过以下方法获得,首先通过Gaussian 03软件进行量化计算,然后通过查阅相关包含类似结构的分子的文献[11]对 Gaussian软件计算结果进行调整。
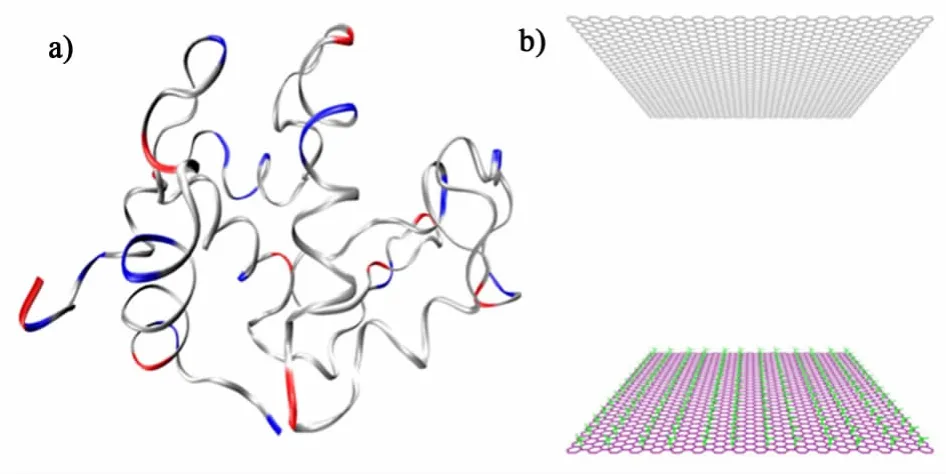
图1 全原子模拟中溶菌酶和色谱孔道模型Fig.1 All-atom model of lysozyme and chromatographic pore structure.a)The lysozyme is shown by a New Cartoon model using the VMD program;b)The chromatographic pore is shown by LINES model using the VMD program
最终,1个溶菌酶分子被放置在带电表面一侧,距离平板约2nm(蛋白质主轴与带电平板平行)。然后将整个盒子居中放入12×12×50nm3的体系中,即在平板上下各加入19nm的真空层以防止周期性边界条件在z方向上对模拟结果的影响。旋转蛋白质的方位以获得不同的初始构象,共进行了3次模拟,同时进行3次将带电介质所带电荷重置为0的空白对照(包括以Cut-off和PME算法为静电作用参数的两组空白对照)模拟。每次模拟时间均为10 ns。
1.2 分子动力学模拟
分子动力学模拟利用GROMACS 4.0.5软件[12,13](http://www.gromacs.org/)完成,使用的力场为 GROMOS96 43A1力场[14-15]。模拟体系为NVT系综,通过Berendsen方法控制温度为298.15 K,时间步长为0.5 ps。势能分析中 LJ势能计算采用Cut-off算法,静电作用力则分别采用Cut-off算法和PME算法,其中Cut-off算法表示在截断半径内按库仑公式计算,在截断半径外则进行补偿,虽然理论而言,越大的截断半径能获到越精确的结果,但是在 Yonetani[2]的研究中发现,使用 Cut-off算法时,截断半径设为1.8nm就能使得水分子出现错误的分层,因此本研究的截断半径根据Gromacs手册[14]设为1.2nm,此算法由于机理简单且运算速度快,广泛用于早期分子动力学模拟;PME算法对截断半径内的静电作用力同样采用库仑计算,但是在计算截断外静电作用力时则是先将体系分为多个小格,后利用三维正向傅里叶变换计算每个格子内的静电作用力,最后直接加和得到静电作用长程部分的能量[16-17],从理论上而言计算结果更加准确,同时模拟所需时间也更长。
所有模拟均使用曙光TC2600刀片服务器完成。文中蛋白质构象图利用VMD软件[18]绘制。
1.3 数据分析方法
1.3.1 最小距离分析
计算蛋白质和带电平板质心之间的距离(dcom)随时间变化,质心距离计算使用的是Gromacs自带的g_dist程序。
1.3.2 二级结构分析
为了描述模拟过程中蛋白质的构象转化,使用GROMACS软件包的do_dssp程序完成其二级结构分析,采用的是Define Secondary Structure of Proteins(DSSP)方法。
1.3.3 分子间势能分析
利用GROMACS软件包的g_energy程序进行势能分析,包括 Lennard-Jones(LJ)势能和库仑势能。计算库仑势能时,Cut-off算法直接利用库仑公式得到所需静电作用能和补偿项相加即所得势能;而PME算法对截断外的长程静电作用力(Coul.recip)采用傅里叶变换进行计算,但是由于算法本身限制,这部分的静电作用力只能以总体值来表示,因此我们采用数学方法对此总体值进行分解,计算出蛋白质和带电平板之间的长程静电作用力,并和截断内作用力加和,进而获得修正库仑势能(Coul.fix)。
2 结果与讨论
2.1 最小距离分析
利用g_dist程序计算对照组蛋白质和带电平板之间的距离变化,如图2所示。
从图2中可以看出,在空白对照组[平板电荷被重置为0,图2a)和图2b)]中,无论是是采用 Cutoff参数或PME参数,蛋白质整体质心变化并不大,蛋白质没有发生明显的位移,仅仅在水的扰动下在其初始位置附近震荡。在平板与蛋白质带同种电荷的情况下,模拟结果出现了差别:首先,蛋白质在两套体系中均发生了较大位移,蛋白质和带电平板之间发生了相互作用,在采用 Cut-off参数的体系中,蛋白质被吸附到平板表面,蛋白质和平板之间最小距离随着时间迅速减小,并稳定在1.5nm左右;而在采用PME参数的体系中,蛋白质被较为快速的排斥远离了带电平板表面,最终稳定在约7nm左右。以上结果直接表明蛋白质在不同参数的体系中会发生不同的行为,说明参数的选取对蛋白质的移动方式有决定作用。根据 Wang[4]的研究结果,在实验过程中并未检测到蛋白质被吸附残留在色谱介质上,因此以Cut-off为静电作用力参数的模拟体系的结果是不符合实际情况的,而以PME为静电作用力参数的模拟结果则和实验结果相符。
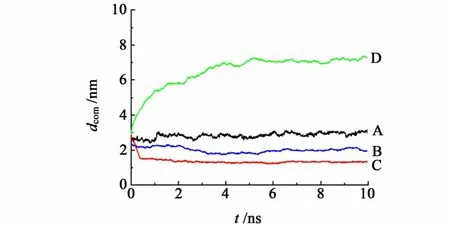
图2 溶菌酶和带电平板之间质心距离随模拟时间的变化Fig.2 The dcom values for protein and the charged surface as a function of simulation time The average of three independent simulation trajectories is used.(A)with the cut-off parameters in control.(B)with the PME parameters in control.(C)with the cut-off parameters in charged system.(D)with the PME parameters in charged system.
2.2 蛋白质二级结构分析
利用DSSP方法确定蛋白质在空白对照组和带电体系中的二级结构随模拟时间的变化,如图3所示。
结果表明,在空白对照中,无论是采用 Cut-off参数或PME参数,经过10 ns的分子动力学模拟,蛋白质的二级结构均保持的较为良好,蛋白质没有发生明显的变性,这也是和实际相吻合的,即蛋白质在纯水环境中不会发生变性。而在带电平板的体系中,情况发生了较为明显的变化,在采用 Cut-off为静电作用力参数的体系中,蛋白质很快失去了大部分的二级结构,且没有自动恢复部分二级结构,说明平板和蛋白质分子之间的作用力使得蛋白质在靠近平板的同时由活性态转变为了变性态,蛋白质被吸附在平板表面且二级结构被破坏;而在采用PME参数的体系中,蛋白质的二级结构虽然也有一定程度的改变,但并未出现各颜色条带长时间消失或增加的情况,这也意味着蛋白质的活性没有受到影响,并且仅有的二级结构变化发生在模拟后期,结合蛋白质质心变化和二级结构变化图可以推测这是由于部分氨基酸残基被带电表面排斥并触碰到了惰性平板造成的。根据 Wang等[4]的研究结果,同电荷色谱介质对蛋白质的复性是促进作用而非抑制作用,因此以Cut-off为静电作用力参数的模拟体系所得结果是不符合实际情况的。
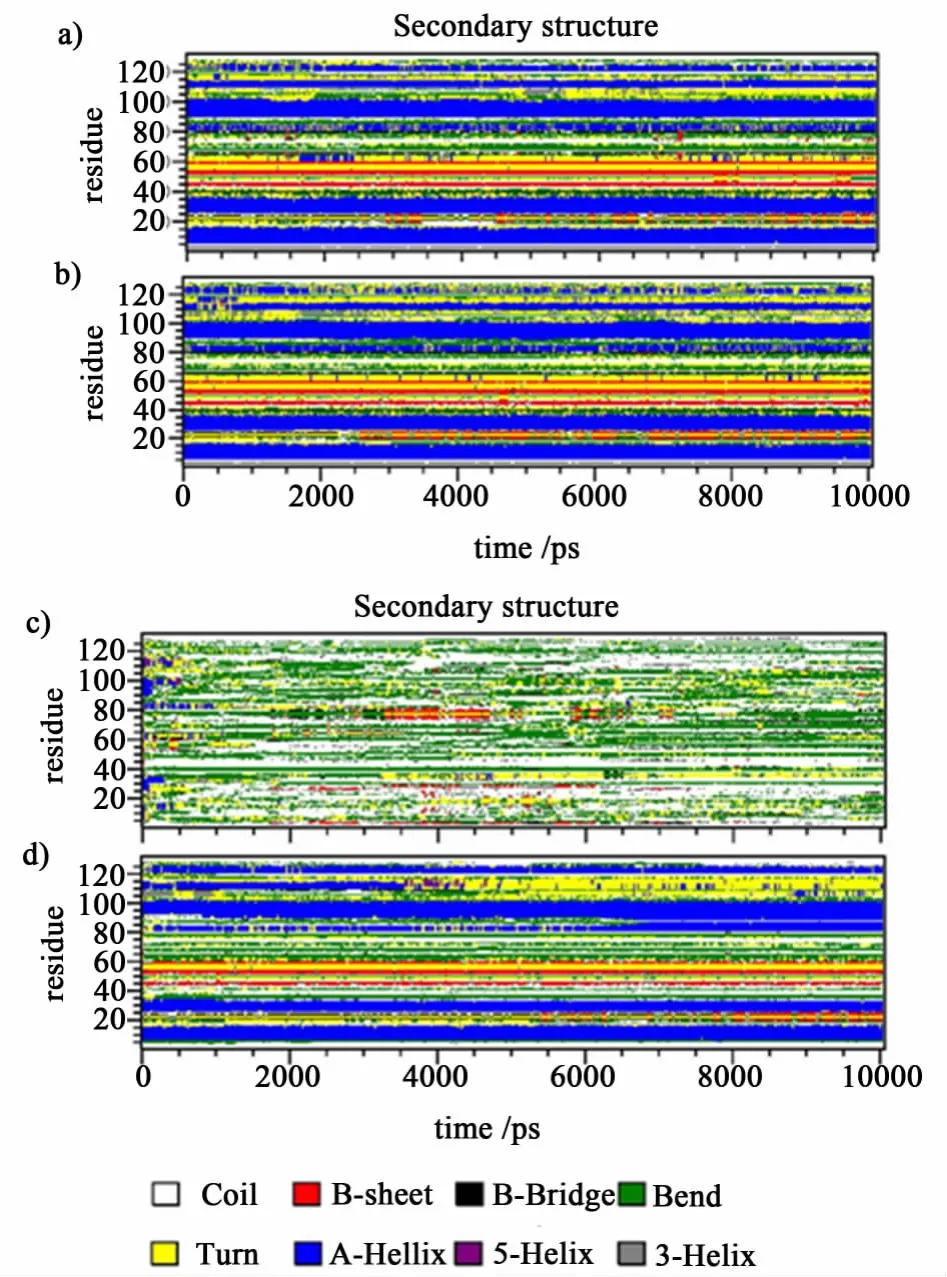
图3 分子动力学模拟中溶菌酶的二级结构变化Fig.3 Secondary structure evolution of lysozyme during the molecular dynamics simulation(A)with cut-off parameters in control.(B)with PME parameters in control.(C)with cut-off parameters in charged system.(D)with PME parameters in charged system.
2.3 分子间势能分析
通过考察蛋白质和表面间相互作用势能分析(在PME体系中所有库仑势能均为修正后的数值),考察蛋白质在不同参数体系中构象及位移出现不同的因素,如图4所示。
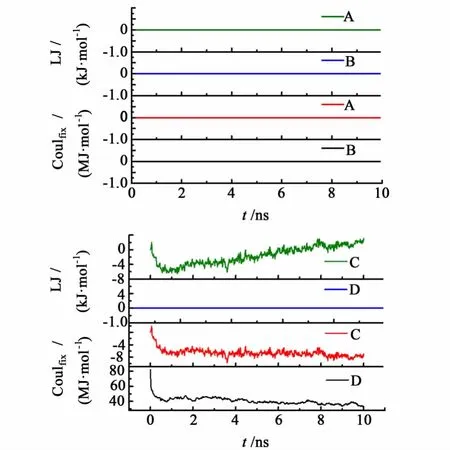
图4 蛋白质和带电平板间作用势能随模拟时间的变化Fig.4 Time evolution of the potential energy between the protein and the charged surface Average of three independent simulation trajectories is used.(A)with cut-off param eters in control.(B)with PME parameters in control.(C)with cut-off parameters in charged system.(D)with PME parameters in charged system.
图4显示,在空白对照中,由于蛋白质和平板之间距离较远,故LJ势能基本为0,由于平板电荷被重置为0,故 Coul势能也为0,也就是说,在这个体系中,蛋白质和平板之间没有相互作用。而在平板带上正电荷之后,在采用 Cut-off参数的体系中,蛋白质和带电平板之间表现为强烈的静电吸引作用(约-8 MJ/mol),且随着时间的延长,越来越多的氨基酸残基和带电平板之间发生了作用,能量呈小幅度波动状态;同时,由于静电吸引导致蛋白质分子和平板之间距离迅速减小,蛋白质和平板之间的LJ势能也表现出来,蛋白质和平板之间出现了亲水作用,然而其数量级仅为 kJ/mol,不起主导作用。而在采用PME参数的体系中,当蛋白处于初始位置时,蛋白质和带电平板之间的库仑作用能很大,但在短时间内库仑作用能下降到较低的水平,蛋白质被迅速的排斥远离了带电表面,然后呈趋向稳定的变化趋势,而自始至终由于蛋白质距离平板较远,蛋白质和带电平板之间未产生明显的LJ势能,这说明蛋白质的排斥是由静电作用力所介导,而且主要是远程静电作用力。
对于采用Cut-off参数处理静电作用力的体系,计算每个氨基酸残基和带电平板之间的作用能表明,较多的带负电的天冬氨酸暴露在溶菌酶表面,使得天冬氨酸和配基发生作用的几率很大,而带正电的氨基酸则较多包埋在氨基酸内部,并且Cut-off算法只计算其截断半径内的静电作用力,对于远程静电作用力则采取简单的补偿法进行修正,这样带正电的氨基酸和配基作用的几率就较小,因而即便溶菌酶分子总体是带的正电荷,仍然发生了蛋白质分子吸附到带电平板的现象。反之,由于PME算法对远程作用力计算的较为精确,溶菌酶分子作为一个带正电的整体能被带电平板排斥远离平板表面,这也是两种算法导致不同模拟结果的最直接原因。但是,需要指出的是,静电作用参数选取对模拟结果的影响和蛋白质本身以及其所处的体系的性质存在很大关系。但随着计算机硬件和软件技术的飞速发展,使用更加精细的算法来进行分子动力学模拟必定是大势所趋。
3 结论
以同电荷介质促进蛋白质复性的实验结果为背景,采用了溶菌酶分子在静电表面的全原子模型,通过使用不同的静电作用力算法来模拟同电荷介质和带电蛋白之间的作用,考察了不同参数下模拟结果的差异及原因。结果表明,在空白对照组中,采用Cut-off算法和PME算法对模拟结果的影响较小,蛋白质没有失去其活性,蛋白质均未发生明显位移。但是,在平板带上同种电荷之后,模拟结果出现了较大差异,采用Cut-off算法的体系中蛋白质被吸附到带电表面,蛋白的结构及活性被强大的静电吸引力破坏,这是与实验结论是不相符的[4];而在采用 PME算法的体系中,蛋白质被远程静电作用力排斥远离了带电平板表面,虽然蛋白质的结构出现过一部分的破坏,但是其骨架结构依旧保存完好,且随着时间的推移,蛋白质的结构出现了一定程度的恢复,这与 Wang[4]的实验结论是相吻合的,而溶菌酶分子的带电特性以及不同算法对长程静电力的处理是造成这个现象的主要因素。因此,对于本体系而言,采用PME算法所得模拟结果明显优于采用 Cut-off算法所得模拟结果。总体而言,本论文通过分子动力学模拟阐述了采取不同静电作用力参数对模拟结果的影响,考察了此过程的微观细节,确定了静电作用力是导致蛋白质被吸附或排斥的主导因素。
[1]Mark P,Nilsson L.Structure and dynamics of liquid water with different long-range interaction truncation and temperature controlmethods in molecular dynamics simulations[J].Journal of Computational Chemistry,2002,23(13):1211-1219
[2]Yonetani Y.A severe artifact in simulation of liquid water using a long cut-off length:Appearance of a strange layer structure[J].Chemical Physics Letters,2005,406(1/3):49-53
[3]Bergdorf M,Peter C.Influence of cut-off truncation and artificial periodicity of electrostatic interactions in molecular simulations of solvated ions:A continuum electrostatics study[J].Journal of Chemical Physics,2003,119(17):9129-9144
[4]Wang G,Dong X,Sun Y.Ion-Exchange resins greatly facilitate refolding of like-charged proteins at high concentrations[J].Biotechnology and Bioengineering,2011,108:1068-1077
[5]白红军,来鲁华.蛋白质相互作用:界面分析,结合自由能计算与相互作用设计 [J].物理化学学报,2010,26(7):1988-1997 Bai Hongjun,Lai Luhua.Protein-Protein interactions:Interface analysis,binding free energy calculation and interaction design[J].Acta Physico-Chimica Sinica,2010,26(7):1988-1997(in Chinese)
[6]Ravichandran S,Madura JD,Talbot J.A brownian dynamics study of the initial stages of hen egg-white lysozyme adsorption at a solid interface[J].The Journal of Physical Chemistry B,2001,105(17):3610-3613
[7]Kubiak-Ossowska K,Mulheran P A.Mechanism of hen egg white lysozyme adsorption on a charged solid surface[J].Langmuir,2010,26(20):15954-15965
[8]Zhang L,Zhao G,Sun Y.Effects of ligand density on hydrophobic charge induction chromatography:Molecular dynamics simulation [J].Journal of Physical Chemistry B,2010,114(6):2203-2211
[9]Diamond R.Real-Space refinement of the structure of hen egg-white lysozyme[J].Journal of Molecular Biology,1974,82(3):371-391
[10]Schuttelkop f A W,Van Aalten D M F.PRODRG:A tool for high-throughput crystallography of protein-ligand complexes[J].Acta Crystallographica Section D:Biological Crystallography,2004,60:1355-1363
[11]Chandrasekhar I,Kastenholz M,Lins R D,et al.A consistent potential energy parameter set for lipids:Dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field [J].European Biophysics Journal with Biophysics Letters,2003,32(1):67-77
[12]Berendsen H J C,Van Der Spoel D,Van Drunen R.GROMACS:A message-passing parallel molecular dynamics implementation [J].Computer Physics Communications,1995,91(1/3):43-56
[13]Lindahl E,Hess B,Van Der Spoel D.GROMACS 3.0:A package formolecular simulation and trajectory analysis[J].Journal of Molecular Modeling,2001,7(8):306-317
[14]Van Gunsteren W,Billeter SR,Eising A A,et al.Biomolecular simulation:The GROMOS96 manual and user guide[B].1996
[15]Schuler L D,Daura X,Van Gunsteren W F.An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase[J].Journal of Computational Chemistry,2001,22(11):1205-1218
[16]Essmann U,Perera L,Berkowitz M L,et al.A smooth particle mesh Ewald method [J].The Journal of Chemical Physics,1995,103(19):8577-8593
[17]Darden T,York D,Pedersen L.Particle mesh Ewald:An N[center-dot]log(N)method for Ewald sums in large systems[J].The Journal of Chemical Physics,1993,98(12):10089-10092
[18]Humphrey W,Dalke A,Schulten K.VMD:Visualmolecular dynamics[J].Journal of molecular graphics,1996,14(1):33-38
