腹泻患者肠道菌群数量变化及E R I C-P C R指纹图谱分析
2014-01-29刘海燕曹海涛常玉梅
刘海燕 曹海涛 常玉梅 徐 鹏
1.解放军第252医院中心实验室,河北保定 071000;2.解放军第252医院医务处,河北保定 071000
腹泻是一种常见病,引起腹泻的原因有多种,大致可分为感染性腹泻和非感染性腹泻,其中感染型腹泻占比较高。感染性腹泻常见的病原菌包括细菌、病毒、真菌、原虫等,其中又以细菌感染为主要感染类型,尤其是在成人腹泻中发生率高。人体菌群失调与腹泻可能存在着某种程度上的因果关系,不同类型的腹泻可能对应着程度不同的菌群数量变化[1-3]。临床上目前大多采用传统的肠道菌群分析方法,需要不同的细菌培养技术,然后进行生化反应鉴定,多种病原菌需要复杂的营养条件,培养操作费时、费力,而且大多数微生物不能很好地培养出来,容易造成假阴性和数量上的偏差,没有成熟的方法完整准确地反映肠道微生物群落的组成情况,采用荧光定量PCR、ERIC-PCR指纹图谱法等分子生态学的方法可有效解决这个问题[4-5]。
1 材料与方法
1.1 样品采集
粪便有白细胞患者(腹泻组)及正常对照者(对照组),粪便样品为2013年7~9月采自解放军第252医院门诊部。腹泻组选自门诊消化科就诊的腹泻患者,对照组选自门诊体检人员,均采自就诊当日,所有样品采集后立即提取基因组DNA,-20℃冷冻保存。所有患者留取粪便标本前均未应用抗生素或微生态制剂。对照组留取粪便标本前2周内无消化道不适症状,且未应用抗生素或微生态制剂。患者资料见表1。

表1 两组一般资料情况
1.2 主要仪器及试剂
粪便基因组DNA提取试剂盒、SYBR Green荧光染料试剂盒均购自北京天根生化科技有限公司,DNA纯化试剂盒、DNA Marker、琼脂糖、PCR 2×PCR Mix购自北京康为世纪生物科技有限公司,实验所需引物为北京天一辉远合成,PCR仪为美国伯乐公司的MyCycler,荧光定量PCR仪为美国AB公司的ABI 7500。
1.3 方法
1.3.1 粪便标本前处理及细菌DNA的抽提 称取2 g粪便加入18 mL无菌PBS,振荡10 min形成粪浆,均匀吸取0.2 g粪浆标本用DNA缓冲液处理,间歇振荡1 min至样本混匀。取上清200 μL,加入1.4 mL缓冲液 GSL,70℃孵育 5 min, 涡旋 15 s,12000 r/min 离心1 min。转移上清液1.2 mL至新的2 mL离心管,加入15 μL蛋白酶K,然后加入缓冲液GB,涡旋片刻后70℃孵育10 min。加入200 μL无水乙醇,涡旋混匀后加入吸附柱,12 000 r/min离心30 s后弃去废液,再用不同浓度漂洗液漂洗3次,待吸附柱在室温放置30 min后,向吸附膜的中间部位悬空滴加50 μL洗脱缓冲液,室温放置5 min,12 000 r/min离心2 min,将溶液收集到离心管中,即为粪便标本中的细菌总DNA。
1.3.2 DNA浓度及纯度的检测 将DNA涡旋片刻,吸取2 μL于Thermo multiskan go酶标仪的微量板的滴样孔处,以吸附缓冲液作为空白对照,每个标本做3个重复,260 nm/280 nm波长处检测吸光度,测定细菌总DNA浓度与纯度,再取5 μL总DNA行1%琼脂糖凝胶电泳,80 V 30 min后于凝胶成像仪观察DNA条带情况。
1.3.3 PCR引物的设计 根据双歧杆菌、乳酸杆菌、大肠埃希菌、肠球菌16S rDNA基因序列,应用引物设计软件 Primer premier 5.0设计并参考文献制订PCR引物。见表2。

表2 肠道菌群与ERIC-PCR所需引物序列
1.3.4 制作标准曲线 取标准菌株冻干粉各0.2 g,用1 mL PBS溶解,振荡混匀后吸取200 μL,按照DNA提取试剂盒标准步骤提取标准菌株基因组DNA,并用相应的引物扩增,扩增产物用PCR产物纯化试剂盒进行纯化后回收,测量纯化后标准菌株DNA OD值,计算双链DNA的浓度,得到PCR片段的拷贝数并换算为各标准品1 μL的拷贝数。实验结果为:双歧杆菌属为 2.3×1014拷贝/μL,乳酸杆菌属为 1.8×1014拷贝/μL,大肠埃希菌属为 5.8×1014拷贝/μL,粪肠球菌为2.6×1014拷贝/μL。然后做10倍系列稀释,使之成为1×102~1×108拷贝/mL,作为定量 PCR 标品,进行实时荧光定量PCR反应,制作标准曲线,其中,横坐标代表不同阳性模板细菌拷贝数的对数值,纵坐标代表出现荧光信号的初始循环数(Ct值)。
1.3.5 荧光定量PCR将待测粪便样品纯化的DNA分别进行3种细菌的SYBR GreenⅠ荧光定量PCR反应,采用20 μL反应体系。荧光染料SYBR premix(2×)9 μL,10 μmol/L 上下游引物各 1 μL, 模板 DNA 2 μL,ddH2O 7 μL,最后加入石蜡油 8 μL。 扩增条件为95℃预变性2 min,95℃变性20 s,双歧杆菌57℃,乳酸杆菌58℃,大肠埃希菌59℃,粪肠球菌60℃退火30 s,68℃延伸 30 s,循环 40 次,74℃后延伸 1 min。 每次反应每个样品做1个复孔,以标准菌株DNA做阳性对照,ddH2O代替DNA模板作为阴性对照。反应完毕后根据标准曲线和样品的Ct值,得出样品中4种细菌的拷贝数。最后根据融解曲线分析PCR产物的特异性。
1.3.6 ERIC-PCR 反应体系为25 μL,DNA模板终浓度为80ng;反应条件:95℃预变性7min,95℃变性1min,52℃退火 1 min,65℃延伸 8 min, 循环 30次,65℃后延伸10 min。反应完毕后于1%琼脂糖凝胶电泳,80 V,30 min,凝胶成像仪检查条带。
1.4 统计学方法
所得数据采用SPSS 17.0统计软件处理,菌群所测结果对数转换后进行统计学分析。正态分布的计量资料用均数±标准差(±s)表示,组间差异采用两独立样本t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 DNA浓度与纯度
DNA浓度检测结果显示,各DNA浓度为200~600 ng/μL,OD260/OD280比值为 1.8~2.2,浓度及纯度可以作为PCR后续使用。部分DNA凝胶电泳结果见图1。

图1 粪便标本DNA电泳结果
2.2 荧光定量PCR
与对照组相比,腹泻组患者粪便中的双歧杆菌属、乳酸杆菌属数量明显减少,差异有统计学意义(P<0.05),大肠埃希菌、肠球菌属数量无明显变化(P>0.05)。两组研究对象的粪便定量结果见表3。
表3 粪便标本细菌定量结果(拷贝/g,±s)

表3 粪便标本细菌定量结果(拷贝/g,±s)
注:与对照组比较,△P<0.05
8.85±0.57 8.28±0.68△8.36±0.45 7.79±0.39△8.41±0.38 8.40±0.50 7.49±0.65 7.56±0.42双歧杆菌属 乳酸杆菌属 大肠埃希菌 肠球菌属对照组腹泻组30 30组别 例数
2.3 ERIC-PCR
ERIC-PCR指纹图谱条带的多少与微生物种群数量呈正相关,条带的敏感程度显示该片段的DNA片段数量。由图2可见,与对照组比较,腹泻组患者ERIC-PCR结果条带明显减少,且小片段居多,多集中在某些特定的片段周围,甚至特定区域的条带多于对照组相应区域,在1500 bp以上的区域条带很少。见图2。
3 讨论
粪检白细胞计数被广泛地用于临床上对感染性和非感染性腹泻的初步诊断,而且对于门诊患者以粪检有无白细胞区分感染性和非感染性腹泻的诊断是可靠的。在腹泻类型中,儿童腹泻以轮状病毒感染居多。本实验主要研究细菌感染类型的腹泻,所以将研究对象设为18岁以上的成年人[6],进行肠道微生态分析。临床上多采用传统的方法进行病原菌的分离与鉴定,需要很长的时间去获得纯培养物,但往往得到的结果却不是很理想,如获得纯培养的细菌比例较少,甚至大量的细菌不能得到培养和鉴定。目前认为,以细菌特异性基因序列分析的分子生物学方法是很可靠的技术,结果理想且时间快,能很好地应用于临床[7-8]。本实验室使用非可培养方法对腹泻组和对照组的肠道菌群进行了分析,用荧光定量PCR方法对4种代表肠道菌群的菌种做了定量研究,用ERIC-PCR法进行肠道菌群总DNA进行扩增,通过样品ERIC条带的分布分析肠道样品中菌群结构的特征,研究对象为肠道内菌群的总和。这两种分子生态学方法均比较快速、简洁,适合在临床上应用推广。
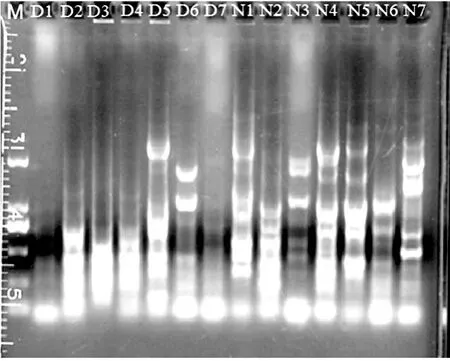
图2 粪便菌群总DNA的ERIC-PCR指纹图谱分析
肠道微生态系统是机体最庞大、最重要的微生态系统。人体肠道内的微生物中,超过99%都是细菌,存活数量大约有10×106个,有500~1000个不同的种类。这些数目庞大的细菌大致可以分为3大类:有益菌、有害菌和中性菌。人体是否健康与肠道内的益生菌群结构息息相关。肠道菌群在长期的进化过程中,通过个体的适应和自然选择,菌群中不同种类之间,菌群与宿主之间,菌群、宿主与环境之间,始终处于动态平衡状态中,形成一个互相依存、相互制约的系统。因此,人体在正常情况下,菌群结构相对稳定,对宿主表现为不致病[9-12]。通常研究肠道菌群主要有大肠埃希菌、肠球菌、双歧杆菌、乳酸杆菌4个种群,当某种外界条件刺激或者机体发生病理改变的时候,菌群结构也发生相应的改变[13-16]。双歧杆菌、乳酸杆菌为有益菌的代表。双歧杆菌、肠球菌为常驻菌群代表。本实验的腹泻研究显示,腹泻患者粪便中的双歧杆菌属、乳酸杆菌属数量明显减少,属于有益菌数量降低,大肠埃希菌、肠球菌属数量无明显变化,整体上说明存在菌群失调现象。菌群失调尤其是益生菌的减少,影响着机体内能量、脂肪和氨基酸的代谢,对维持机体代谢平衡起着重要作用。菌群失调和多种原因的腹泻有着密切关系。SYBR GreenⅠ是一种可以和双链DNA结合的荧光染料,在定量PCR过程中,由于不断地与新的PCR产物结合,形成的荧光信号也越来越强,通过标准曲线的方法可以得到定量结果,相对于探针法简单、节约经费,在引物特异性较好的情况下可以准确得到混合菌群中各菌株的数量,应用于菌群的定量研究。
ERIC序列是首先在肠道细菌基因组中发现的非编码保守重复序列,被广泛应用于种内或者种间各类微生物的鉴定,也有越来越多的研究者用来分析混合菌群的群落结构,得到能够反映菌群组成差异的重复性好而又稳定的图谱。ERIC序列作为一种长引物RAPD技术,应用到研究生态系统中微生物群落结构,具有重复性好、简便、快速和灵敏度高的特点,可以有效地同时分析不同生态系统的结构差异以及动态地检测同一生态系统微生物群落的结构变化。ERIC-PCR指纹图谱技术已经广泛应用于混合微生物的研究中,在研究土壤、活性污泥、肠道菌群等复杂微生物菌群中可以分析出微生物群落的组成特征[17-19]。本研究通过ERIC-PCR指纹图谱技术分析粪检有白细胞的腹泻患者及健康对照者肠道样品,比较腹泻与健康个体肠道菌群结构的整体差异,研究腹泻对肠道菌群结构的影响。结果显示,粪检白细胞的患者肠道菌群结构与正常人肠道菌群存在着很大的差异。首先得到的结论是腹泻患者肠道内扩增条带范围小而集中在较小片段,暗示着该组菌群多样性明显减少。
本研究用DNA荧光定量PCR法和ERIC-PCR指纹图谱技术对腹泻人群与对照组进行检测,比较肠道微生物菌群结构的变化情况。这两种方法直接检测微生物的基因组DNA,用DNA的种类组成代表细菌的种类组成,可以比较客观、全面地反映微生物群落结构中优势菌群的结构变化,不仅克服了传统方法只能检测少部分种类的局限性,而且缩短了分析时间,提高了分析精度,还能够对大量样品进行并行分析,使得系统检测肠道菌群区系结构随环境变化而变化的规律成为可能。
[1]Arkkila P,Mattila E,Anttila VJ.Fecal transfusion as treatment of Clostridium difficile infection[J].Duodecim,2013,129(16):1671-1679.
[2]Sachdev AH,Pimentel M.Gastrointestinal bacterial overgrowth:pathogenesis and clinical significance [J].Therapeutic Advances in Chronic Disease,2013,4 (5):223-231.
[3]Israeli E,Shoenfeld Y.Harnessing nature for treating infectious and autoimmune diseases:good and bad bacteria[J].Harefuah,2013,152(4):188-189.
[4]Vondrakova L,Pazlarova J,Demnerova K.Detection,identification and quantification of Campylobacter jejuni,coli and lari in food matrices all at once using multiplex qPCR[J].Gut Pathogens,2014,6(1):12.
[5]Chen L,Li Y,Yu Y.Soil bacterial and fungal community successions under the stress of chlorpyrifos application and molecular characterization of chlorpyrifos-degrading isolates using ERIC-PCR[J].Journal of Zhejiang University:Science B,2014,15(4):322-332.
[6]刘文林.123例腹泻患者大便检验结果分析[J].按摩与康复医学,2012,3(36):478-479.
[7]Rigsbee L,Agans R,Shankar V,et al.Quantitative profiling of gut microbiota of children with diarrhea-predominant irritable bowel syndrome[J].Am J Gastroenterol,2012,107(11):1740-1751.
[8]王成龙,苏东华,刘佼佼,等.不同基因型变异链球菌临床分离株的分离和鉴定[J].华西口腔医学杂志,2013,31(1):80-85.
[9]高兴,辛文文,高姗,等.11种(株)食源性细菌基因芯片检测方法的建立[J].生物技术通报,2013(12):123-128.
[10]黄遵锡.胃肠道微生物及其分子生态学技术研究进展[J].微生物学通报,2014,41(1):136-145.
[11]Michael B,Thomas C.Metabolic diversity of the intestinal microbiota:implications for health and disease[J].J Nutr,2007,137(3 suppl 2):751-755.
[12]Backhed F,Dina H,Wang T,et al.The gut microbiota as an environmental factor that reguLates fat storage[J].Proc Natl Acad Sci USA,2004,101(44):15718-15723.
[13]Robosky LC,Wells DF,Egnash LA,et al.Metabonomic identification of two distinct phenotypes in Sprague-Dawley (Crl:CD(SD)) rats[J].Toxicol Sci,2005,87(1):277-284.
[14]Clayton TA.Metabolic differences underlying two distinct rat urinary phenotypes,a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism[J].FEBS Letters,2012,586(7):956-961.
[15]Williams HRT,Cox IJ,Walker DG,et al.Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn's disease[J].BMC Gastroenterology,2010,10(1):108.
[16]David MR,Andrew JS,Steve PH,et al.Matrix metalloproteinase 9 contributes to gut microbe homeostasis in a model of infectious colitis[J].BMC Microbiology,2012,12:105.
[17]He L,Xie X,Wang C.16S rDNA Identification and phylogenetic analysis of six strains of lactobacillus from female genital tract[J].Chinese Journal of General Practice,2013,4:71.
[18]He GZ,Deng SX,Qian N,et al.Intestinal microbial community diversity between healthy and orally infected rabbit with Entamoeba histolytica by ERIC-PCR[J].Parasitol Res,2012,111(3):1123-1126.
[19]Wang FQ,Zhang P,Jang HL.Gut Microbial diversity in rat model induced by Rhubarb [J].Digestive Diseases and Sciences,2012,57(11):2856-2862.
