HPLC-MS-MS同位素内标法测定火腿中14 种喹诺酮类残留量
2014-01-18祝子铜徐佳文雷美康章应俊
祝子铜,徐佳文,雷美康,彭 芳,章应俊
(衢州出入境检验检疫局,浙江 衢州 324002)
HPLC-MS-MS同位素内标法测定火腿中14 种喹诺酮类残留量
祝子铜,徐佳文,雷美康,彭 芳,章应俊
(衢州出入境检验检疫局,浙江 衢州 324002)
建立同时测定火腿中14 种喹诺酮类化合物高效液相色谱串联质谱检测方法。样品采用乙腈提取,乙腈饱和正己烷脱脂,经MAX固相萃取小柱净化,采用高效液相色谱串联质谱多反应监测正离子模式测定,使用同位素内标定量。14 种喹诺酮类检出限为0.6 μg/kg。方法平均回收率在70.0%~115.0%之间,相对标准偏差在1.2%~15.6%之间。该方法前处理简便快速、选择性好、灵敏度高,可作为日常的检测方法在实验室中使用。
火腿;高效液相色谱串联质谱;同位素内标法;喹诺酮类
喹诺酮类药物是近年来研究较多的一类抗菌药,被广泛用于人和动物疾病的治疗,但该类药物在动物体内的残留可通过食物链对人体健康构成危害[1]。目前,我国农业部以及世界卫生组织、美国、欧盟、日本等国家和组织都将该类药物列入限制使用的兽药名单中,并制定出相关的最高残留限量[2]。出口至美国、加拿大等国家的动物源性食品喹诺酮类检测项目达15 项之多,出口至日本的检测项目也有10 项。因此,为确保出口产品的安全,打破国外的技术壁垒,有必要建立一个简便快速的动物源性食品中喹诺酮类药物的检测方法。
目前对于喹诺酮类药物残留的报道很多,主要方法有高效液相色谱法[3-7]、高效液相色谱串联质谱(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS-MS)法[8-15]、毛细管电泳法[16-17]、气相色谱-质谱法[18]。本研究在前人研究的基础上,针对国标方法中采用正己烷净化后[19],还存在着很强的基质干扰,对净化条件进行了优化,采用正己烷脱脂后,调节pH值至11,使用MAX固相萃取柱(solid phase extraction,SPE)净化,三重四极杆-质谱同位素内标法分析火腿中14 种喹诺酮类药物。该方法前处理简便快速、选择性好、灵敏度高、基质干扰小,完全能够满足世界各国对火腿中喹诺酮类药物残留的限量要求,本方法可在实验室日常检测中使用。
1 材料与方法
1.1 材料与试剂
MAX SPE(150 mg/6 mL) 上海安谱公司;甲醇、乙腈、正己烷(均为色谱纯) 德国默克公司;甲酸、冰乙酸(均为色谱纯) 阿拉丁试剂(上海)有限公司;氢氧化钠(分析纯) 上海国药集团化学试剂有限公司;标准物质:氧氟沙星、诺氟沙星、恩诺沙星、环丙沙星、单诺沙星、奥比沙星、沙拉沙星、马波沙星、依诺沙星、洛美沙星、双氟沙星、恶喹酸、萘啶酸、氟甲喹 德国Dr Ehrenstorfer公司;依诺沙星-D8、氧氟沙星-D3、诺氟沙星-D5、环丙沙星-D8、恩诺沙星-D5、双氟沙星-D4、沙拉沙星-D8 德国Witega公司。
1.2 仪器与设备
1260-6460型液相色谱串联四极杆质谱仪 美国安捷伦科技公司;12位固相萃取装置 德国CNW公司;Biofuse primo R离心机、Barnstead Nanopure超纯水仪 美国赛默飞公司;MS3漩涡振荡器、T18BASIC ULTRA-TURRAX均质器 德国IKA公司;R-210旋转蒸发仪 瑞士Büchi公司;N-EVAPTM111氮吹仪 美国Organomation公司。
1.3 方法
1.3.1 标准溶液的配制
标准储备液:分别精密称取适量的喹诺酮类标准品和喹诺酮类氘代内标标准品,加入一定量的甲酸溶解标准品,再用甲醇稀释并定容,其中恶喹酸先用氨水溶解,再用甲醇稀释并定容,配制成0.1 mg/mL的标准储备液。该溶液在4 ℃的冰箱中保存。
中间标准储备液:分别准确吸取标准储备液,用甲醇逐级稀释成1.0 μg/mL混合溶液。
标准工作溶液:吸取不同体积的中间标准储备液,用体积分数0.15%甲酸溶液配成2、5、10、20、50 ng/mL标准工作溶液,其中内标为20 ng/mL,现配现用。
1.3.2 样品前处理
准确称取火腿样品2 g(精确至0.01 g)于50 mL离心管中,加入内标溶液,加入20 mL乙腈,均质2 min,涡旋提取1 min,以6 000 r/min离心5 min,取出上清液于150 mL分液漏斗中,再加入20 mL乙腈,重复上述操作,合并提取液于分液漏斗中,加入25 mL乙腈饱和正己烷,振摇2 min,静置待两相分层,取出乙腈层于鸡心瓶中,在45 ℃条件下旋蒸至近干,加入5 mL乙腈饱和的正己烷,涡旋后转移至50 mL离心管中,再加入10 mL超纯水溶解残渣,转移至50 mL离心管中,以6 000 r/min离心5 min,弃去正己烷层,水相用0.1 mol/L氢氧化钠溶液调节pH值至11,待净化。
MAX SPE柱用5 mL甲醇、5 mL水活化平衡,将样品溶液倒入SPE小柱中,依次用5 mL体积分数5%氨水、5 mL甲醇淋洗,用9 mL 体积分数5%甲酸-甲醇进行洗脱,接收全部洗脱液,50 ℃条件下用氮气吹干,加入1 mL体积分数10%甲醇溶液(含体积分数0.15%甲酸),超声1 min,涡旋混合1 min,过0.22 μm滤膜,待分析。
1.3.3 HPLC条件
色谱柱:Zorbax XDB-C18(4.6 mm×150 mm,5 μm);流动相:A相为体积分数0.15%甲酸溶液;B相为甲醇;柱温40 ℃;流速0.4 mL/min;进样量20 μL;HPLC流速及梯度洗脱见表1。
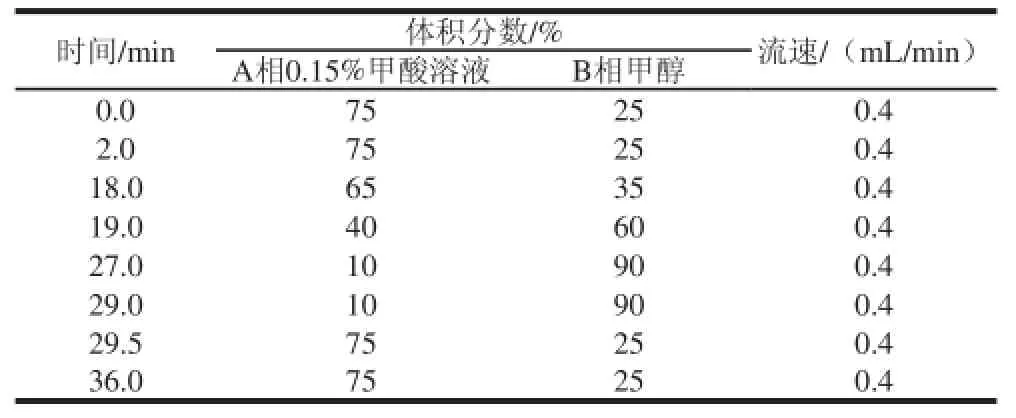
表1 液相色谱流速及梯度洗脱程序Table1 HPLC flow rate and gradient elution program
1.3.4 MS参数
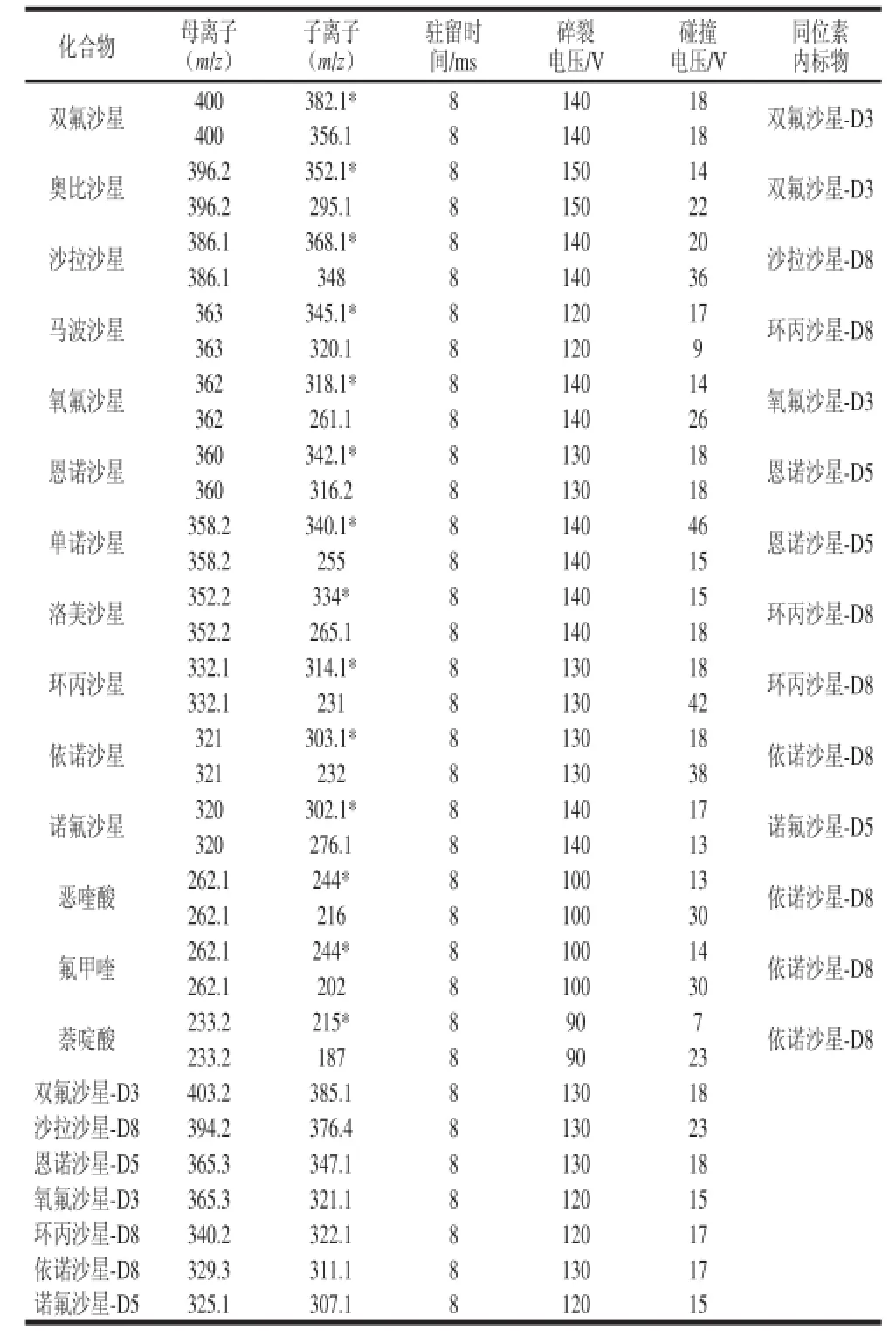
表2 14 种喹诺酮类MS测定条件Table2 Optimized spectrometric parameters for 14 quinolone residues
电喷雾离子源,正离子模式;干燥气温度350 ℃;干燥气流速5 L/min;喷雾气压力45 psi;鞘气温度400 ℃;鞘气流速:12 L/min;毛细管电压4 000 V;多反应监测扫描采集参数见表2。
2 结果与分析
2.1 提取条件优化
喹诺酮类分为哌嗪喹诺酮和酸性喹诺酮。喹诺酮既有羧基又有哌嗪环,因此在水溶液中具有酸性和碱性2 种性质。喹诺酮类药物的这种特殊特性决定其可溶于多种溶液而能从生物样品中提取出来。喹诺酮类药物常用的提取溶剂有酸性溶液、碱性溶液、纯有机溶剂。本研究比较了乙腈-甲酸、乙腈-乙酸、乙腈、甲醇-乙酸、甲醇-甲酸、甲醇等提取溶液进行提取的效果,发现乙腈、乙腈-甲酸体系作为提取溶液对大部分喹诺酮类的提取效果较好。但是乙腈-甲酸作为提取试剂,提取后溶液比较浑浊,干扰物较多,在HPLC-MS-MS分析中基质抑制效应很强,其次,随着甲酸的加入,萘啶酸、氟甲喹、恶喹酸这几个化合物提取效率降低,该结果与文献报道的乙酸乙酯、丙酮和酸化甲醇主要用于提取酸性喹诺酮,乙腈、酸化乙腈适合提取哌嗪类喹诺酮一致。因此,本实验采用纯乙腈作为提取溶液进行提取。
2.2 净化条件优化
2.2.1 SPE柱的选择
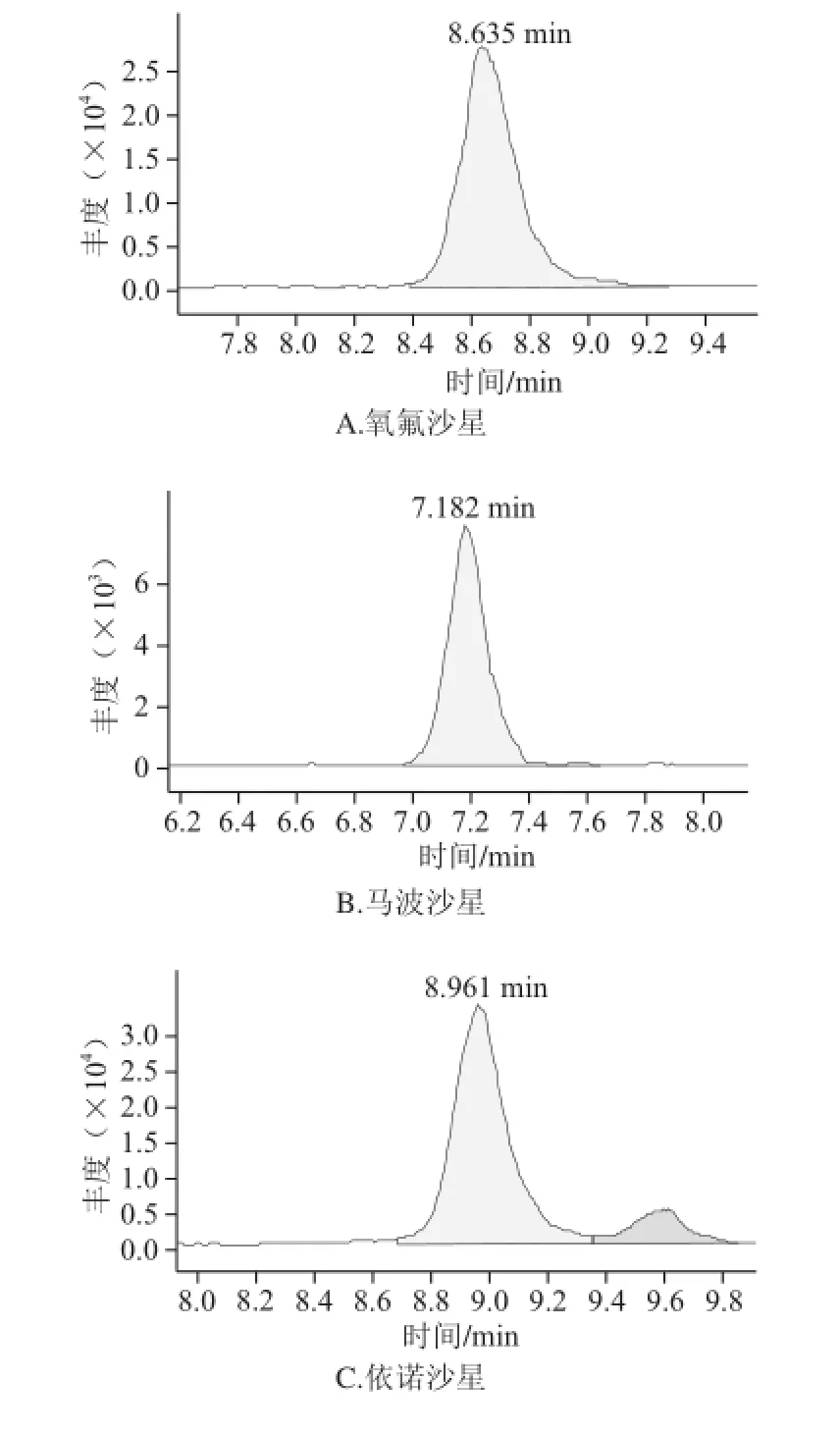
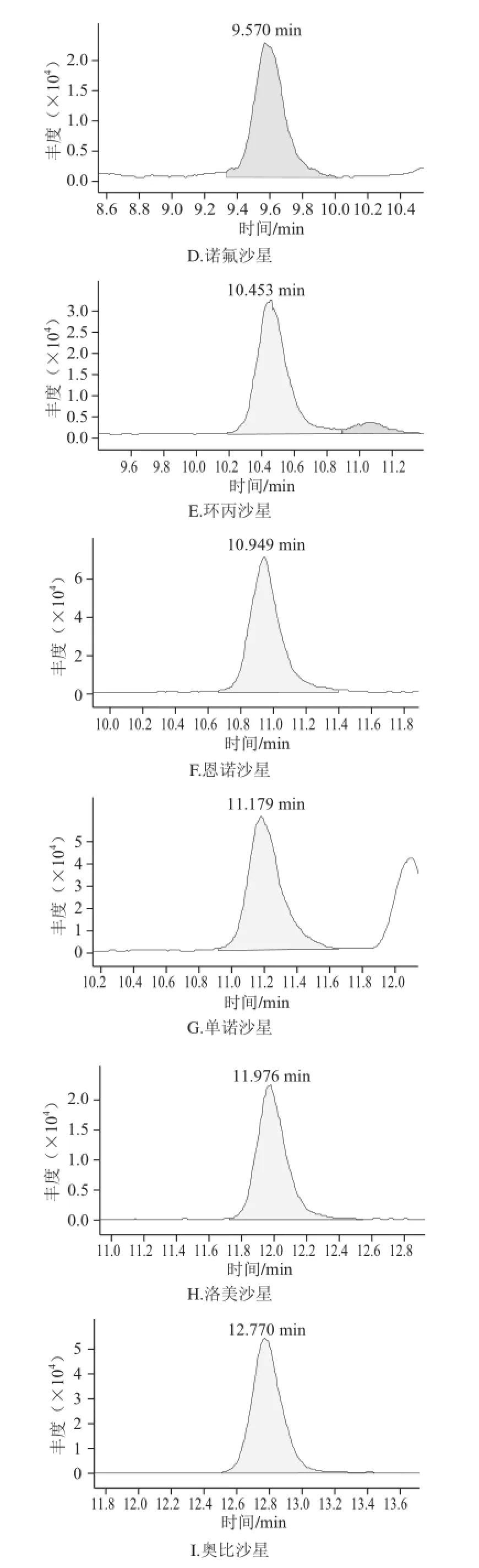

图1 MAX固相萃取柱净化后火腿样品中喹诺酮类的选择离子色谱图Fig.1 SRM chromatograms of 14 QNS purified with MAX SPE column
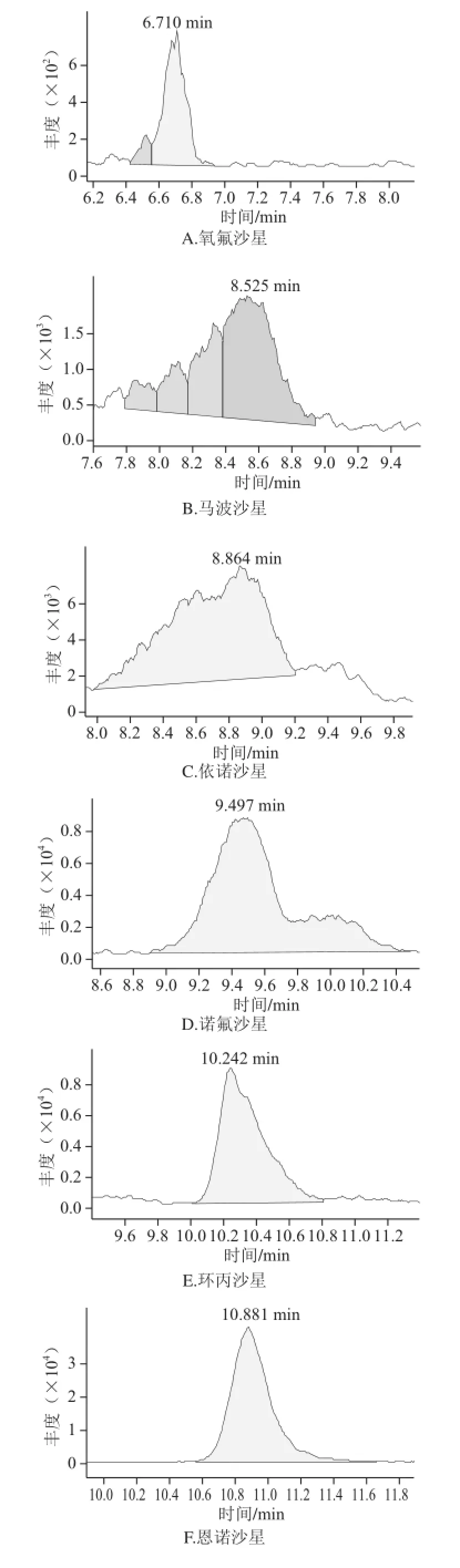
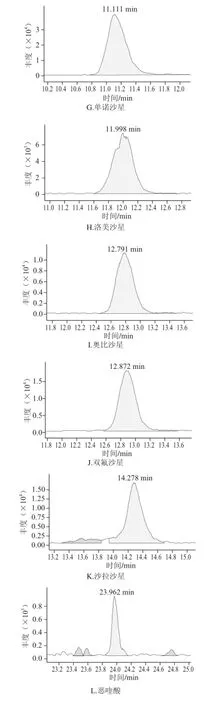
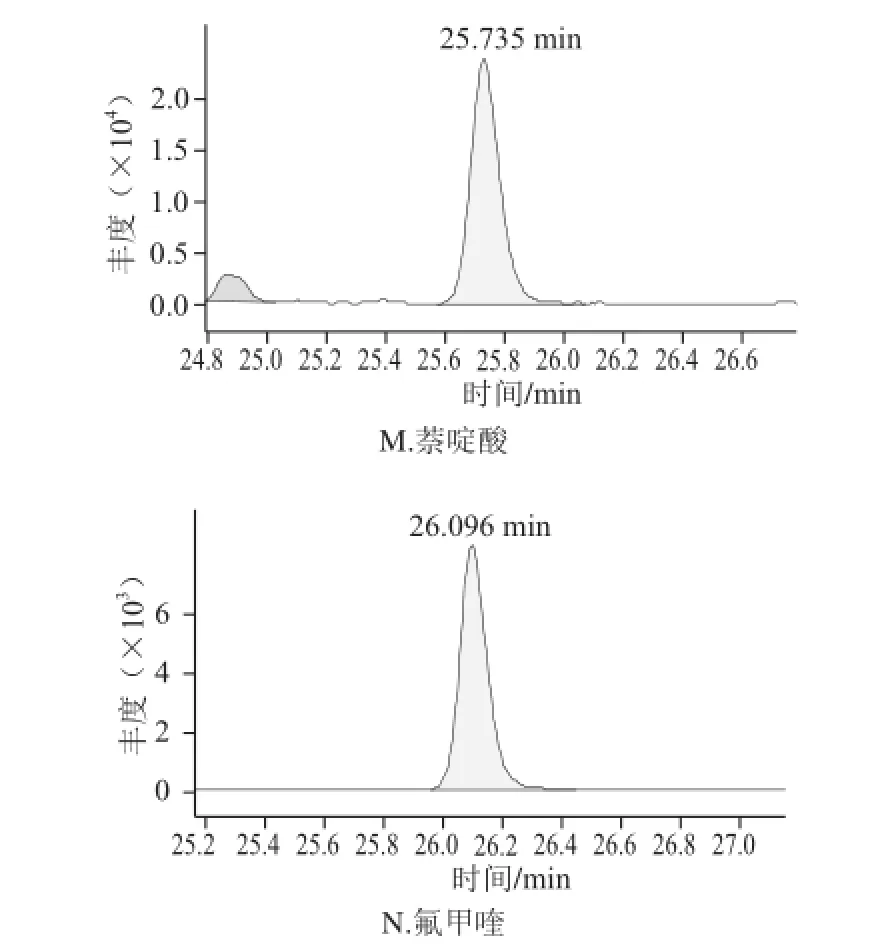
图2 正己烷净化后火腿样品中喹诺酮类的选择离子色谱图Fig.2 SRM chromatograms of 14 QNS purified with hexane
由于火腿样品基质复杂,特别是火腿中含有大量的脂肪成分,在HPLC-MS-MS分析中存在严重的基质抑制作用。要同时分析多种喹诺酮类药物残留,仅仅通过采用正己烷去脂一步净化不能有效地去除干扰物质。因此,本研究采用正己烷先去脂,再过SPE小柱对样品进行进一步净化。目前已经开发了各种各样通用的SPE方法,如果样品前处理所采用的原理和液相色谱分析的原理是正交不相关的,则通常可以得到最好的净化结果。正交的方法有时又被称为二维方法,它采用2 个完全不同的分离模式或保留机理来极大地提高分离能力[20]。举例来说,如果样品前处理后进行反相HPLC分析,则选择性最好的SPE方法是离子交换作用。喹诺酮类具有酸性和碱性2 种性质,在水溶液中以离子态的形式存在。因此,本研究采用MAX SPE小柱进行净化。由图1和图2比较后可以得出,经过MAX SPE小柱净化后,各化合物受基质抑制影响减弱、色谱峰形明显改善、灵敏度提高。
2.2.2 上样液pH值的选择
用氢氧化钠溶液将超纯水分别调至pH值为9.0、10.0、11.0、12.0,然后添加相同量的标准溶液,进行回收率实验。总体而言,14 种喹诺酮类药物在4 个pH值条件下都有较高的回收率,而pH 11.0时大部分药物回收率最高。pH 9.0~10.0时,环丙沙星、氧氟沙星、奥比沙星、双氟沙星回收率较低。因此,本研究选用pH 11.0为上样液的最佳pH值。
2.2.3 洗脱液的选择
配制甲酸-甲醇(5∶95,V/V)溶液,分别取6、7、8、9、10 mL进行洗脱,并计算回收率。实验结果显示6~8 mL甲酸-甲醇溶液不能完全将喹诺酮类药物洗脱下来。当洗脱液体积达到9 mL时,喹诺酮类能够被完全洗脱。因此,本实验选择使用9 mL甲酸-甲醇溶液作为洗脱液。
2.3 色谱条件优化
色谱柱是色谱分析的核心,同一样品在不同填料的色谱柱上分离效果不同,通过Agilent Zorbax XDB-C18柱(4.6 mm×150 mm,5 μm)、Agilent Zorbax SB-C18柱(2.1 mm×100 mm,1.8 μm)、Agilent Eclipse XDB-C18柱(2.1 mm×50 mm,1.8 μm)3 种色谱柱考察了15 种化合物的分离度和峰形,Agilent Eclipse XDB-C18色谱柱,喹诺酮类化合物峰形较差,Agilent Zorbax XDB-C18、Agilent Zorbax SB-C18色谱柱都能够达到基线分离大部分喹诺酮类物质,考虑到火腿样品基质复杂,且实验室样品量较大,而5 μm色谱柱抗污染能力较强。因此,本实验选择Agilent Zorbax XDB-C18色谱柱作为日常分析柱。
2.4 质谱参数建立
本研究采用HPLC进样方式在正离子模式下对待测化合物的标准溶液分别进行一级全扫描,确定准分子离子峰[M+H]+,再分别以待测化合物的准分子离子为母离子进行碎裂,进行二级子离子扫描,选择2 个特征子离子,分别选择信噪比高、峰形好、干扰小的离子对作为定量离子对。对于喹诺酮类,二级质谱图都有明显的[M+H—H2O]+和[M+H—CO2]+峰,同时由于喹啉环的6、7位被氟原子与哌嗪基取代,因而与丢失HF和因派嗪基团上C—N键与C—C键断裂而造成H转移形成新基团所产生的峰也十分突出。
2.5 内标物选择
应用HPLC-MS-MS技术分析样品中痕量兽药残留时,基质效应是时常遇到的现象,基质效应可导致测试结果重复性差,定量结果难以保证[21]。同位素内标法是目前HPLC-MS-MS分析中经常使用的一种定量技术,由于同位素内标物结构与目标分析物一致,受基质影响程度相同,在质谱中的碎裂行为相似。因此,同位素内标法可有效地降低基质效应的影响,提高定量的准确性。本方法选用目前能够购买到的依诺沙星-D8、氧氟沙星-D3、诺氟沙星-D5、环丙沙星-D8、恩诺沙星-D5、双氟沙星-D4、沙拉沙星-D8作为相对应化合物喹诺酮类的内标物,而奥比沙星、司帕沙星、马波沙星、单诺沙星、洛美沙星、恶喹酸、氟甲喹和萘啶酸没有相对应的同位素内标。本研究分别使用上述7 种同位素内标物作为奥比沙星的内标并作校准曲线,用上述校准曲线对实际加标样品进行计算,选取回收率结果与实际加标最接近的曲线作为校准曲线,该校准曲线的内标物为双氟沙星-D4。司帕沙星、马波沙星、单诺沙星、洛美沙星、恶喹酸、氟甲喹和萘啶酸的内标依据上述方法选取。14 种喹诺酮类对应的内标物见表2。
2.6 方法的线性关系、检出限和定量限
在本实验所确定的条件下,以待测物质的质量浓度为横坐标,以标准品与同位素内标物峰面积比值y为纵坐标,分别绘制14 种喹诺酮类标准溶液工作曲线,由表3可知,14 种喹诺酮类在2~20 ng/mL范围内呈良好的线性关系。该方法的检出限和定量限由空白样品添加回收实验获得,以3 倍信噪比为检出限、10 倍信噪比为定量限,14 种喹诺酮类兽药的检出限和定量限分别为0.6 μg/kg和2.0 μg/kg。
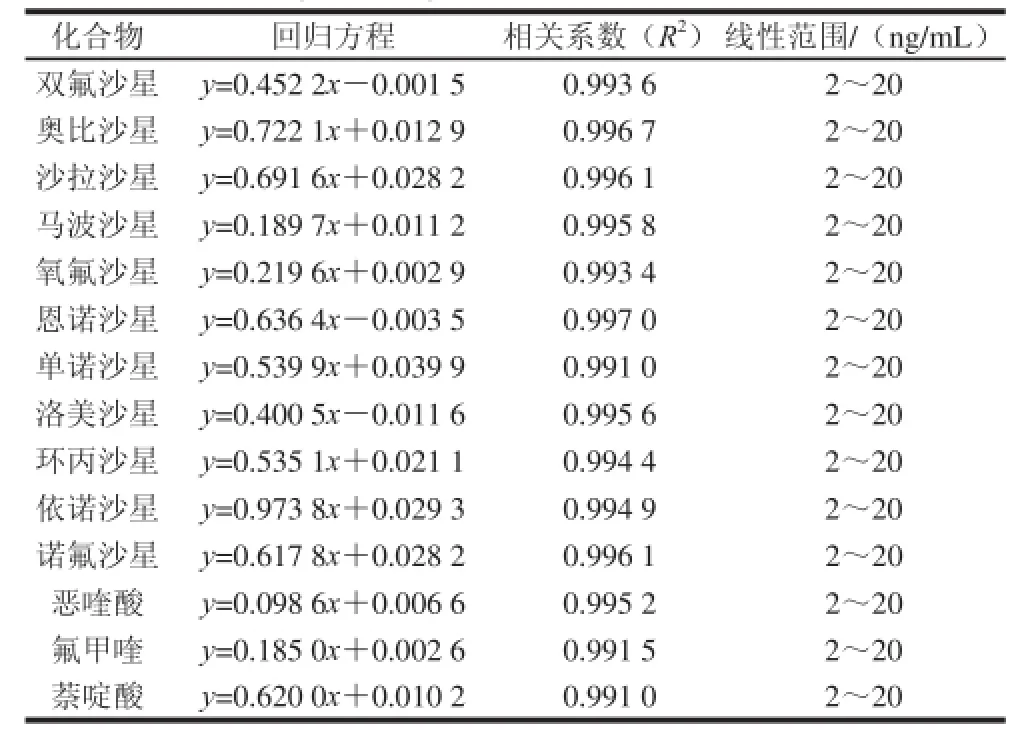
表3 14 种喹诺酮类的线性方程及线性范围Table3 Linear ranges and regression equations forof 14 quinolone residues
2.7 精密度和回收率
为测定本方法的精密度和回收率,实验选用空白火腿为样品基质,添加不同量的喹诺酮类混合标准溶液,进行加标回收实验,每个化合物做2.0、5.0、10.0 μg/kg 3 个水平的添加,每个添加水平均做6 平行样,其结果见表4。由表4可以看出,14 种喹诺酮类方法平均回收率在70.0%~115.0%之间,相对标准偏差在1.2%~15.6%之间。方法的准确度和精密度均符合残留分析的要求。
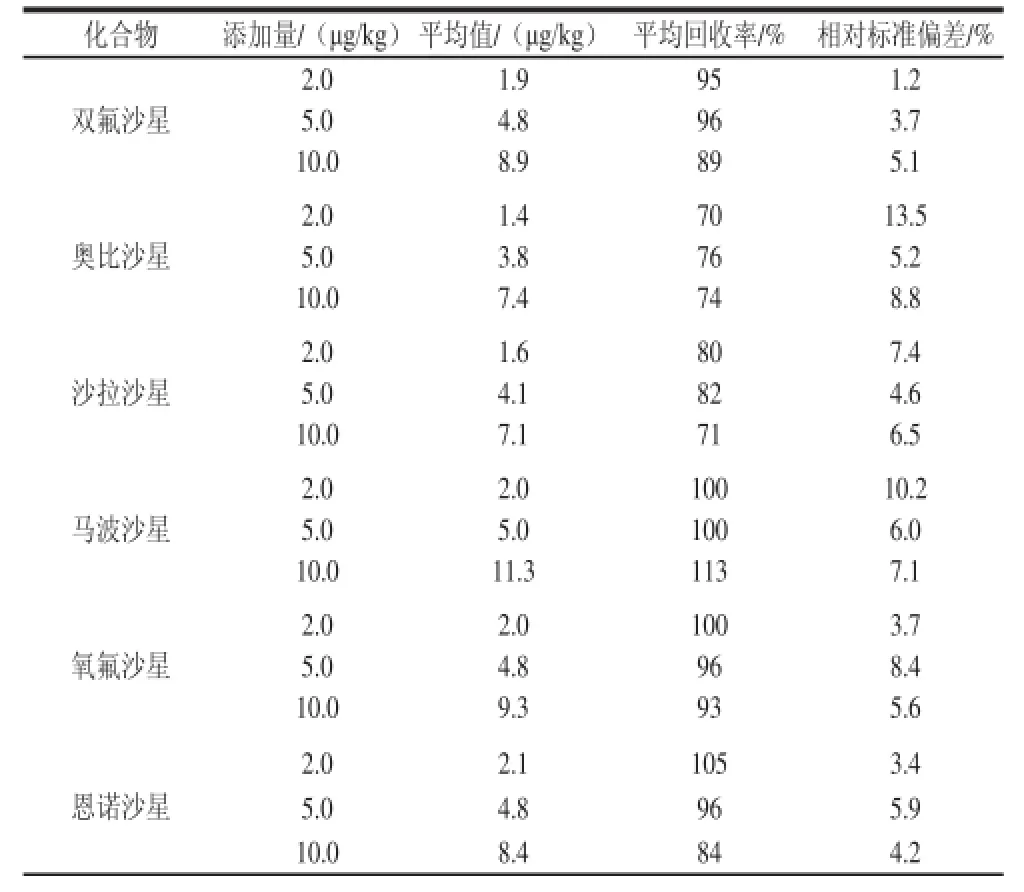
表4 回收率与精密度实验结果(n=6)Table4 Mean recovery rates and precision for 6 replicate determinations (n=6)
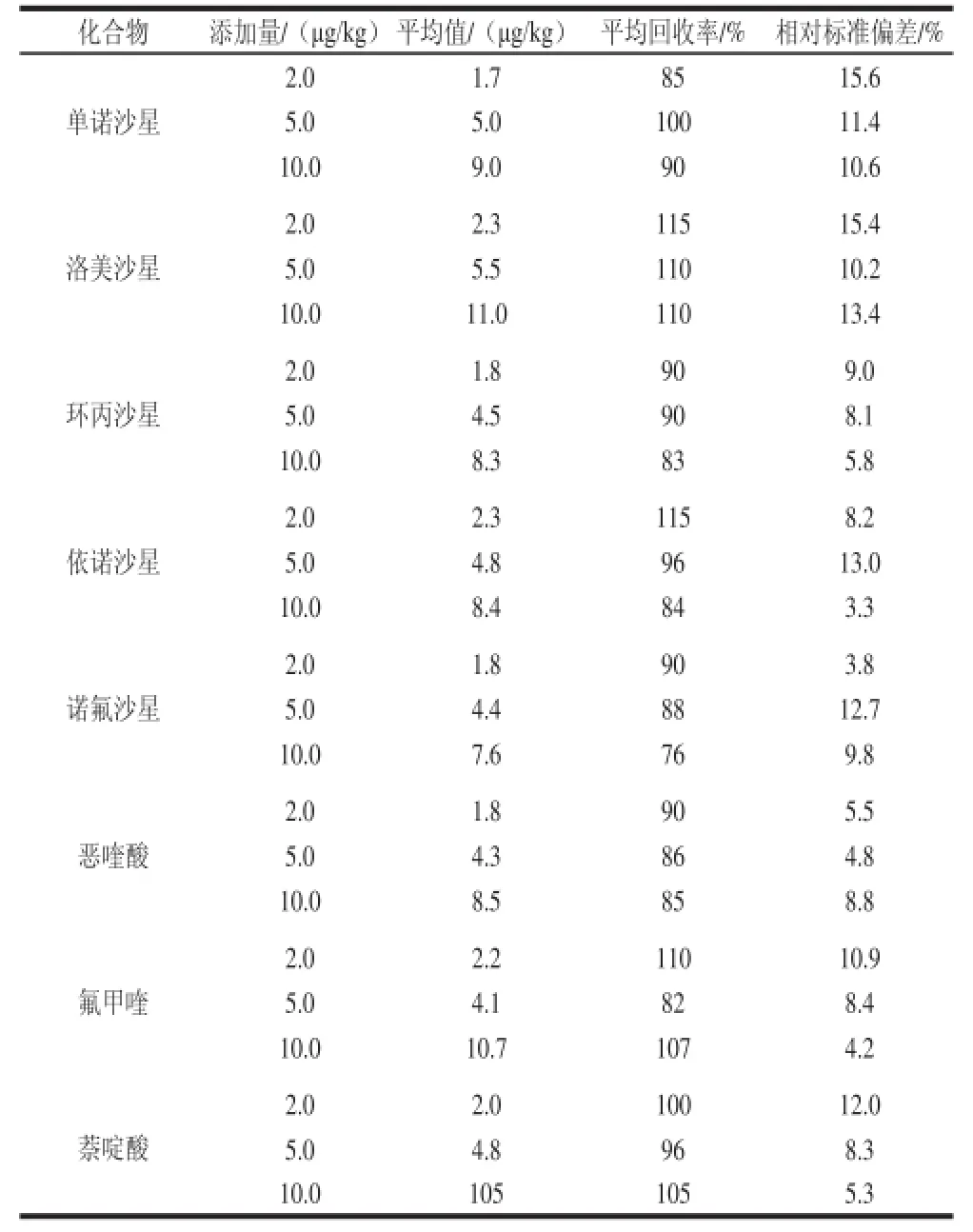
续表4
3 结 论
本研究建立火腿中14 种喹诺酮类残留检测的HPLCMS-MS分析方法。通过对HPLC条件和MS条件各项参数的优化,以及采用MAX SPE柱对样品提取液净化和同位素内标法补偿基质效应,使得方法的灵敏度、准确度和精密度均能满足兽药残留分析方面的要求。本方法可用于实验室日常检测,用此方法对100多份样品进行了检测,均取得了满意的结果。
[1] 李俊锁, 邱月明, 王超. 兽药残留分析[M]. 上海: 上海科学技术出版社, 2002.
[2] 吴永宁, 周宗灿, 王绪卿. 现代食品安全科学[M]. 北京: 化学工业出版社, 2003: 153-182.
[3] 惠芸华, 张晓玲, 金高娃, 等. 高效液相色谱法测定水产品中9 种氟喹诺酮类药物残留[J]. 海洋渔业, 2010, 32(2): 204-210.
[4] 钱卓真, 苏秀华, 魏博娟, 等. 高效液相色谱法同时测定水产品中6种喹诺酮类药物的残留[J]. 食品科学, 2010, 31(6): 185-189.
[5] 占春瑞, 温志海, 卜延刚, 等. 肌肉中多种喹诺酮类兽药残留量的高效液相色谱测定研究[J]. 食品科学, 2005, 26(10): 172-176.
[6] ROUDAUT B, YORKE J C. High-performance liquid chromatographic method with fluorescence detection for the screening and quantification of oxolinic acid, flumequine and sarafloxacin in fish[J]. Journal of Chromatography B, 2002, 780(2): 481-485.
[7] 董琳琳, 刘艳华, 汪霞, 等. 反相高效液相色谱法同时测定4 种氟喹诺酮类药物在鸡可食性组织中的残留[J]. 色谱, 2005, 23(3): 285-288.
[8] 厉文辉, 史亚利, 高立红, 等. 加速溶剂萃取-高效液相色谱-串联质谱法同时检测鱼肉中喹诺酮、磺胺与大环内酯类抗生素[J]. 分析测试学报, 2010, 29(10): 987-992.
[9] 曹慧, 陈小珍, 朱岩, 等. QuEChERS-超高效液相色谱-串联质谱技术同时测定乳制品中磺胺类和喹诺酮类抗生素残留[J]. 食品科技, 2013, 38(6): 323-329.
[10] 彭涛, 雍炜, 安娟, 等. 反相高效液相色谱/质谱法同时测定鸡肉中5种喹诺酮药物残留[J]. 分析化学, 2006, 34(增刊): 10-14.
[11] 黄优生, 刘波平, 朱筱玲, 等. 高效液相-串联质谱法快速测定鱼肉中4 种氟喹诺酮类药物残留[J]. 食品科学, 2010, 31(2): 127-130.
[12] TOUSSA1NT B, CHEDIN M, BORDIN G, et a1. Determination of (fluro) quinolone antibiotic residues in pig kidney using liquid chromatography-tandem mass spectrometry: partⅠ. Laboratoryvalidated method[J]. Journal of Chromatography A, 2005, 1088(1/2): 32-39.
[13] 丁涛, 沈东旭, 徐锦忠, 等. 高效液相-串联质谱法测定蜂蜜中残留的19 种喹诺酮类药物[J]. 色谱, 2009, 27(1): 34-38.
[14] 岳振峰, 谢丽琪, 陈小霞. 牛奶中16 种喹诺酮类药物残留量的高效液相色谱-串联质谱法测定[J]. 分析测试学报, 2008, 27(3): 240-243.
[15] 马建民, 夏曦, 李晓薇, 等. 阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13种喹诺酮类药物[J]. 中国食品卫生杂志, 2013, 25(3): 249-253.
[16] BARRON D, JIMENEZ-LOZANO E, BAILAC S. Determination of difloxacin and sarafloxacin in chicken muscle using solid-phase extraction and capillary electrophoresis[J]. Journal of Chromatography B, 2002, 767(2): 313-319.
[17] HORSTKOTTER C, JIMENEZ-LOZANO E, BARRON D. Determination of residues of enrofloxacin and its metabolite ciprofloxacin in chicken muscle by capillary electrophoresis using laser-induced fluorescence detection[J]. Electrophoresis, 2002, 23(17): 3078-3083.
[18] CHEN G, SCHNEIDER M J. A rapid spectrofluorometric screening method for enrofloxacin in chicken muscle[J]. Journal of Agricultural and Food Chemistry, 2003, 51(11): 3249-3253.
[19] 山东出入境检验检疫局. GB/T 20366—2006 动物源产品中喹诺酮类残留量的测定: 液相色谱-串联质谱法[S]. 北京: 中国标准出版社, 2006.
[20] Waters. LC/MS分析中的“离子抑制”分析综述[EB/OL]. [2014-03-12]. http:∥www.waters.com/waters/library.html.
[21] 王立琦, 贺利民, 曾振灵, 等. 液相-串联质谱检测兽药残留中的基质效应研究进展[J]. 质谱学报, 2011, 32(6): 321-332.
Determination of 14 Quinolone Residues in Ham by HPLC-MS-MS with Isotopic Internal Standard
ZHU Zi-tong, XU Jia-wen, LEI Mei-kang, PENG Fang, ZHANG Ying-jun
(Quzhou Entry-Exit Inspection and Quarantine Bureau, Quzhou 324002, China)
A high performance liquid chromatography-tandem mass spectrometry (HPLC-MS-MS) was developed for simultaneous determination of 14 quinolone residues (QNS) in ham. Ham sample was extracted with acetonitrile, defatted with hexane saturated with acetonitrile and cleaned up by MAX solid phase extraction. The mass spectrometer was operated in the positive ion mode using select reaction monitoring for qualitative and quantitative analysis. Isotope internal standard was used for quantitative analysis. The limit of detection of the method was 0.6 μg/kg. Mean recovery rates for 14 QNS in a negative ham sample spiked at three levels were between 70% and 115%, with RSD ranging from 1.2% to 15.6%. This method proved to be of simplicity, high sensitivity and good selectivity. It has been used for routine test in our laboratory.
ham; high performance liquid chromatography-tandem mass spectrometry (HPLC-MS-MS); isotopic internal standard method; quinolone (QNS)
TS207.3
A
1002-6630(2014)20-0258-07
10.7506/spkx1002-6630-201420051
2014-01-08
祝子铜(1985—),男,工程师,硕士,主要从事动物源产品中抗生素残留检测研究。E-mail:zztzzt1124@163.com
